processQFeatures App
Léopold Guyot
Loïc Guille
15 July 2026
Source:vignettes/processQFeatures.Rmd
processQFeatures.RmdThis app can be used once the data from
importQFeatures() have been downloaded. In order to do
that, unzip the folder downloaded, and load the RDS that contain the
QFeatures object in your R environment or pass directly the path as an
argument when using processQFeatures(). You can also
directly start the application processQFeatures() and load
the RDS into the application.
Start the app
Parameters of the Shiny application:
qfeatures(optional): a QFeatures object. This can be either a path to an RDS file or a QFeatures object already loaded in your R session.prefilledSteps(optional): defines the different steps of the workflow used to analyse the QFeatures object. Accepted values aresampleFiltering,featureFiltering,normalisation,missingValuesFeatures,missingValuesSamples,zeroToNA,logTransform,imputation,aggregation, andjoin. The suggested workflow set as the default value issampleFiltering,featureFiltering,missingValuesFeatures,missingValuesSamples,normalisation,aggregation,join, andaggregation.initialSets(optional): sets to use for the analysis of the QFeatures object. The default isseq_along(qfeatures).
The app can be started without any parameter, in that case the app will start by asking you to load an RDS containing a QFeatures object. Once loaded you can adjust the sets needed for the analysis of the object.
Workflow configuration
A predefined workflow is defined by default with the argument
prefilledSteps. If prefilledSteps was modified
when the application was launched, this change will be taken into
account.
The page contains a brief explanation of each step. To modify the
workflow, drag and drop the different steps to be executed on the
QFeatures object. Once the workflow is configured, click on
Confirm Current Workflow.
If the default workflow is the one you want to use, or if you have
already specified the desired workflow in the
prefilledSteps parameter, simply click on
Step 1.
Sample/Feature Filtering
Pre-Filtering Metrics section
pre-Filtering metrics section
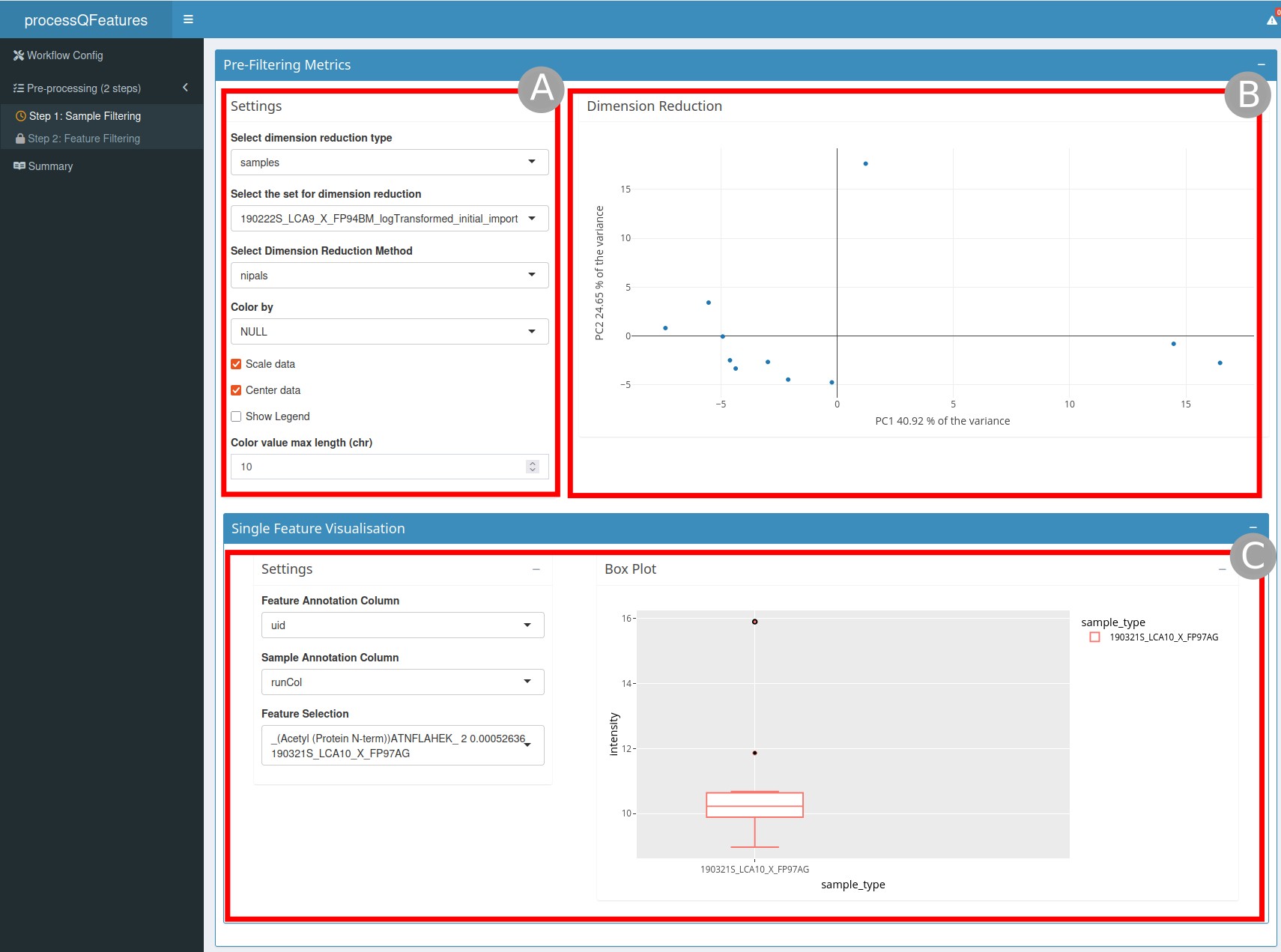
The Pre-Filtering Metrics section (Figure 1) is composed
of two different parts:
a dimension reduction graph (
B) on the right and the settings (A) to customize this graph on the left. For example, you can choose the dimensionality reduction method or type.a single feature visualization (
C) where you can display a boxplot of a selected feature split by sample annotation.
Filtering section
filtering section
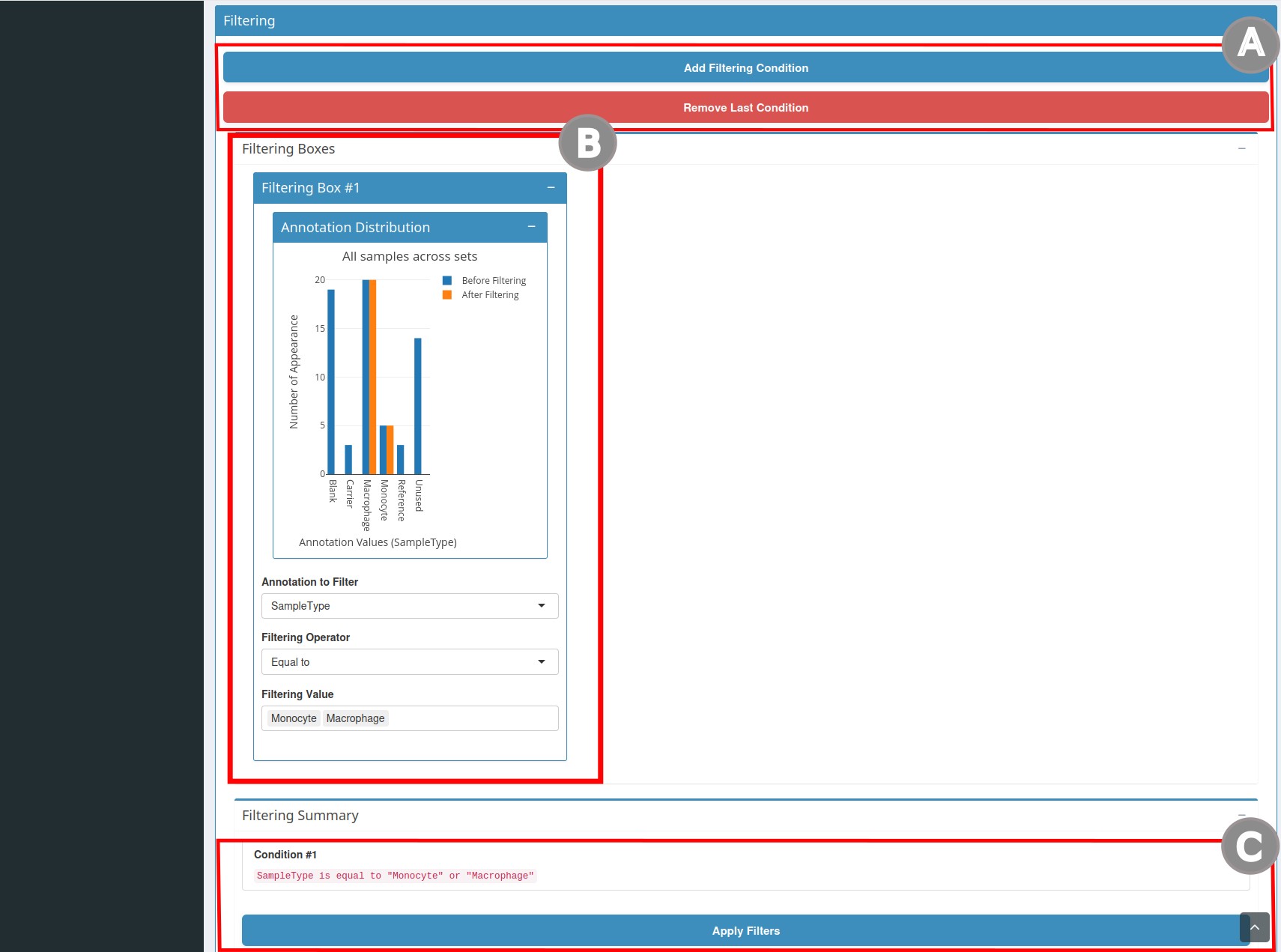
- The second section is
Filtering(Figure 2). To create a condition, clickAdd Filtering Condition(A). Clicking this button will open the filtering boxes (B). In this box, you can add filtering conditions. Once you have chosen the annotation and the value to filter, the number of cells that will be filtered is updated dynamically. You will also find a summary of the selected conditions. Once the filters are chosen, clickApply Filters(C). If a condition is unnecessary, you can also click onRemove Last Condition(A).
Post-filtering Metrics section
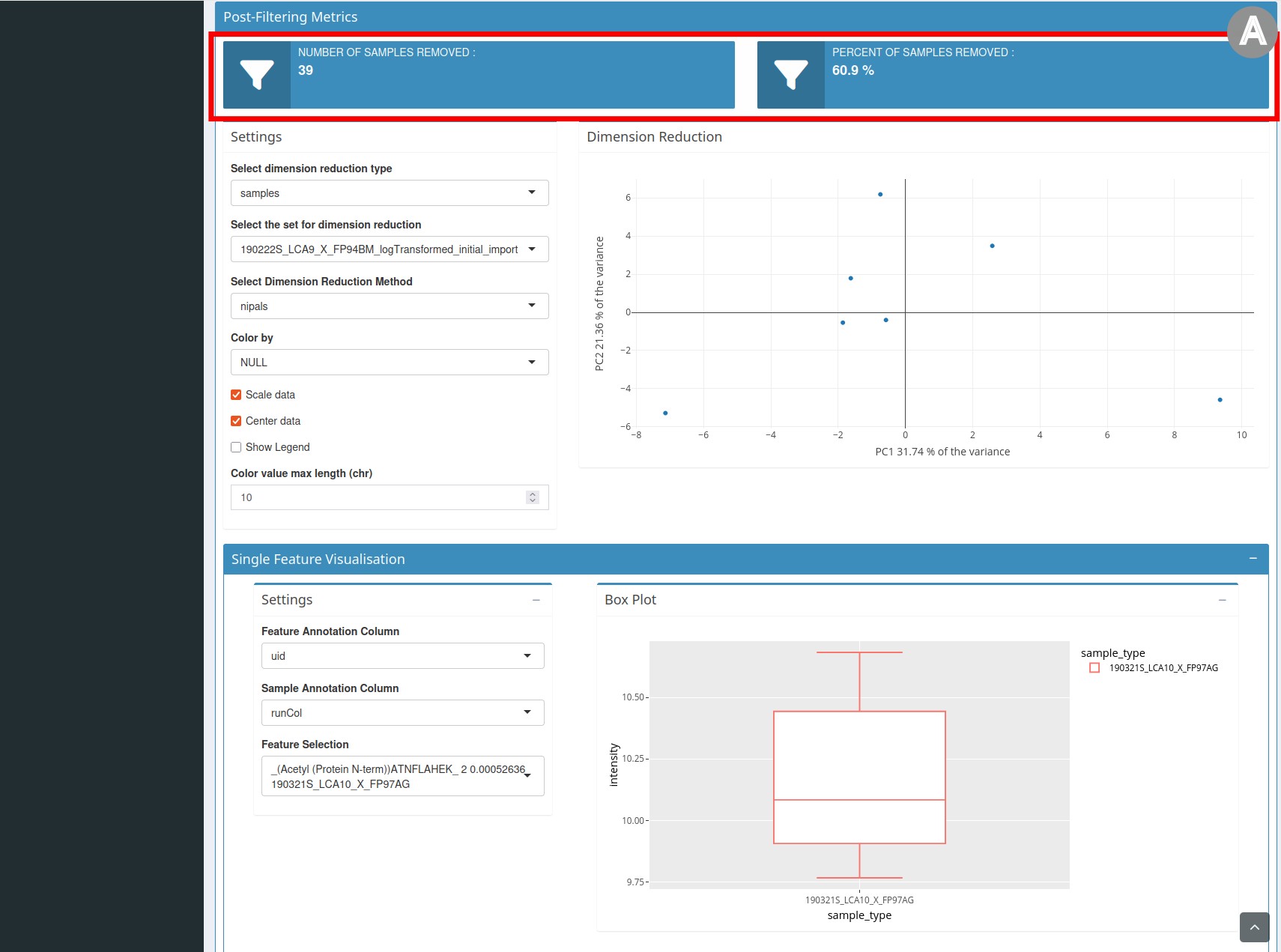
post-Filtering metrics section
- The third section, entitled
Post-Filtering Metrics(Figure 3), repeats the elements of thePre-Filtering Metricssection, but the graph presents the data after filtering. It also contains statistics on the number and percentage of samples/features removed.
Once the filtering has been done, save the object by clicking the
button Save the processed sets.
Filtering missing values by samples/features
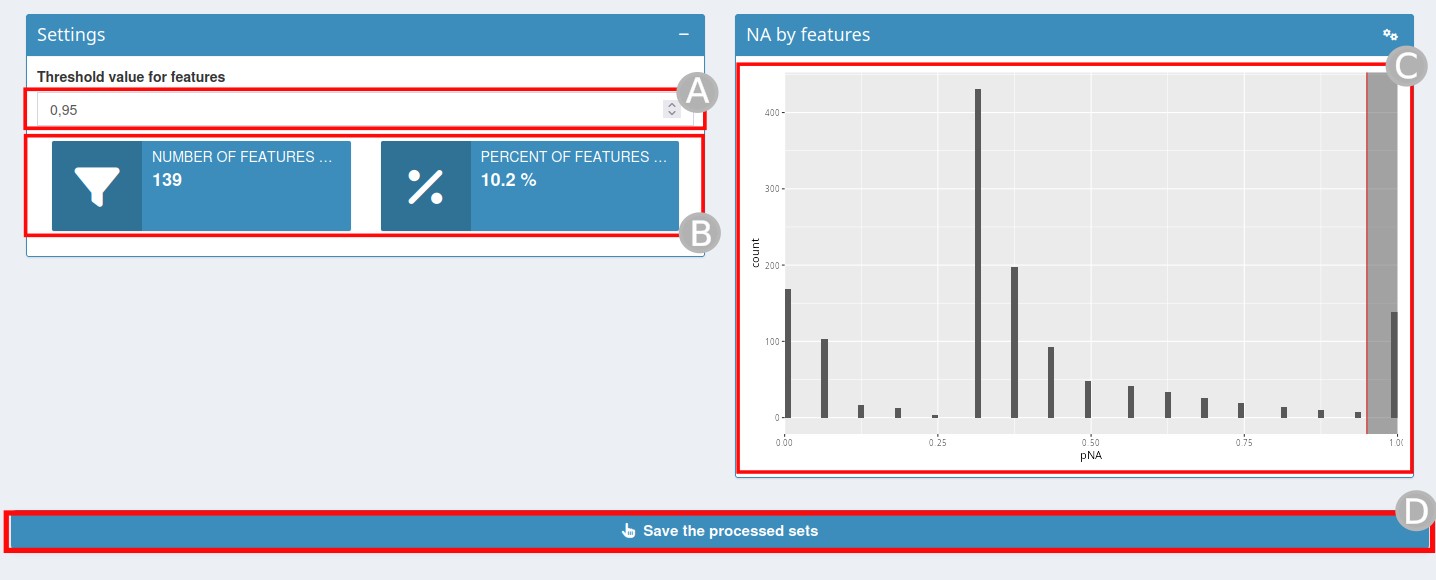
Filtering missing values by samples/features page
Missing values can be very numerous in certain proteomics experiments and need to be dealt with carefully.
In this step, you will need to define a threshold for the percentage
of missing values (NA) beyond which a feature/sample should be removed
(A). By changing this value, the statistics relating to the
number and percentage of features/samples removed (B) and
the associated graph (C) will be automatically updated.
Once the threshold is defined, click on
Save the processed sets (D).
Normalisation
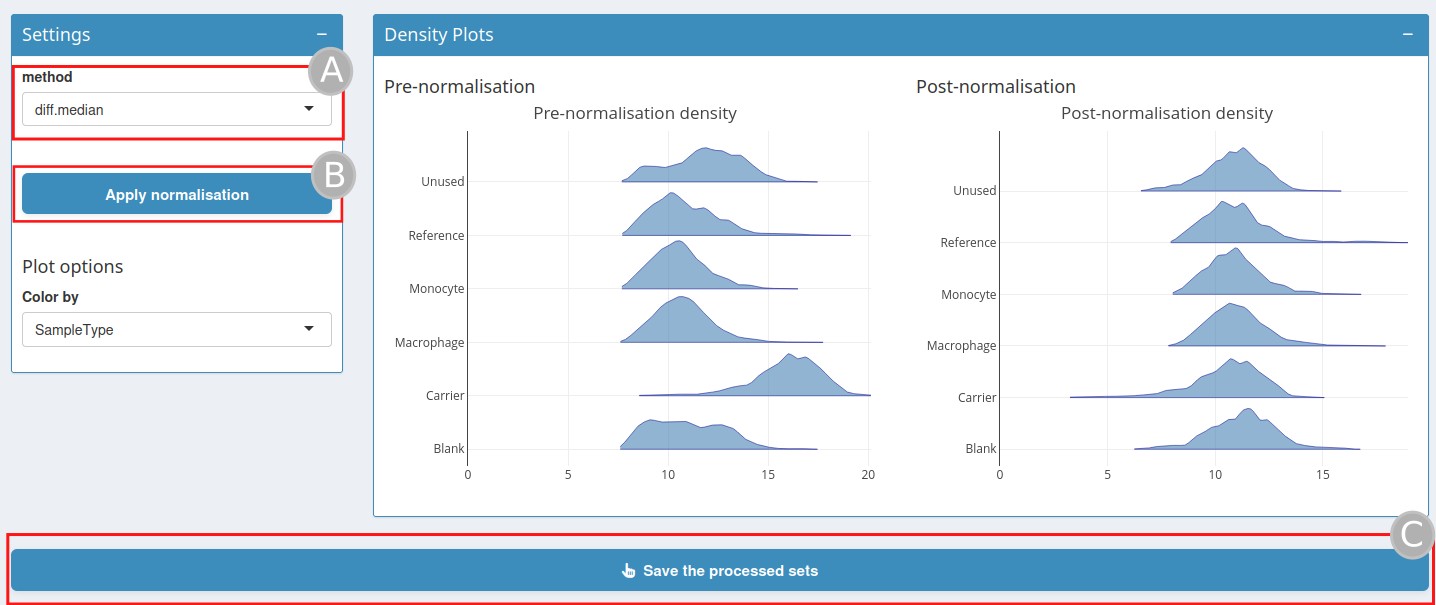
Normalization page
QFeatures objects can be normalised in this step. Several
normalisation methods are available (A).
sumandmax, where each feature’s intensity is divided by the maximum or the sum of the feature, respectively. These two methods are applied along the features (rows).center.meanandcenter.mediancentre the respective sample (column) intensities by subtracting the respective column means or medians.div.meananddiv.mediandivide by the column means or medians.diff.mediancentres all samples (columns) so that they all match the grand median by subtracting the respective column median differences from the grand median.Using
quantilesorquantiles.robustapplies (robust) quantile normalisation, as implemented inpreprocessCore::normalize.quantiles()andpreprocessCore::normalize.quantiles.robust().vsnuses thevsn::vsn2()function. Note that the latter also glog-transforms the intensities. See the respective manuals for more details and function arguments.
Once the normalisation method is selected, click on
Apply normalisation (B); the density graph
after normalisation will be displayed. Click on
Save the processed sets (C).
Zero to NA
Zero to NA page
This step converts all zero intensities into missing values (NA) in the selected sets.
Log Transformation
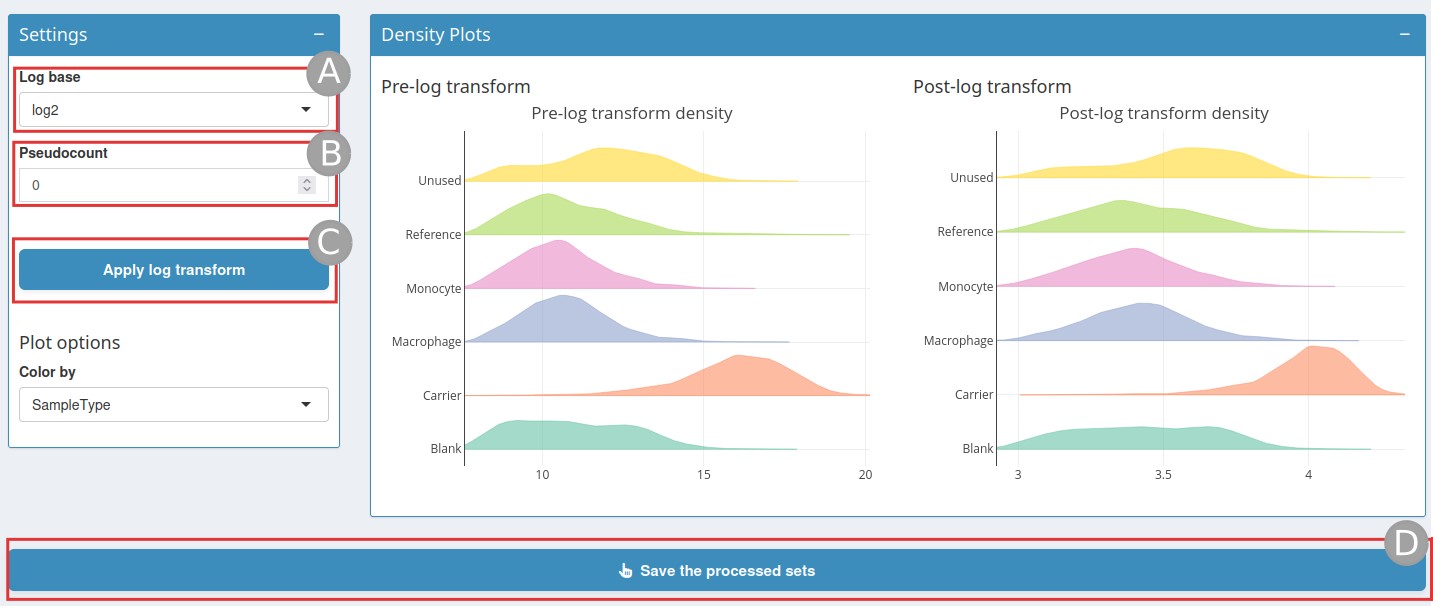
Log transformation page
When analysing continuous data using parametric methods (such as t-test or linear models), it is often necessary to log-transform the data.
The log base is set to log2 by default, but can also be
set to log10 or ln (A). A
pseudocount value can be added (B) to handle zero values in
the data before applying the logarithm. Then click on
Apply log transform (C); this will display a
density plot after log transformation. Once this is done, do not forget
to click on Save the processed sets (D).
Imputation
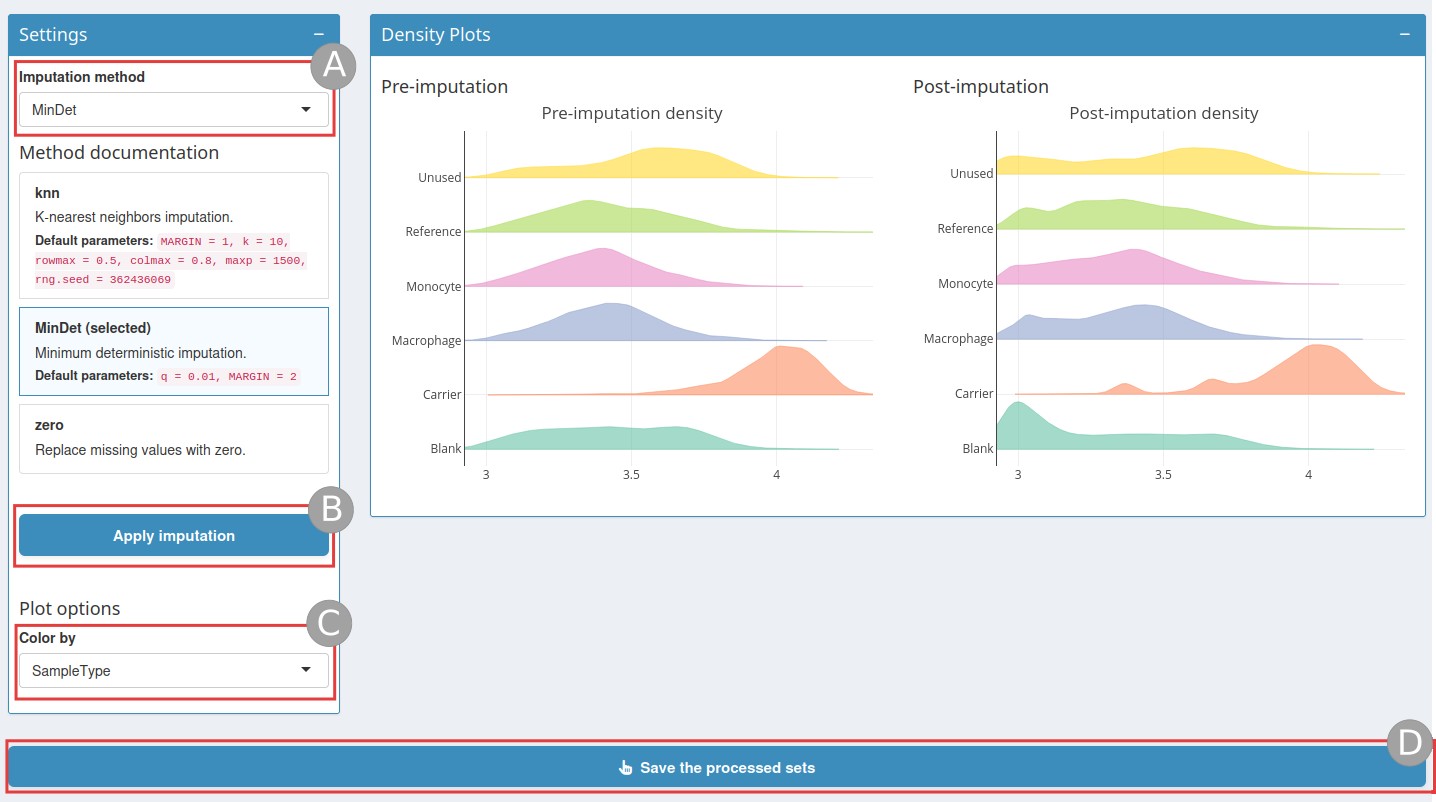
Imputation page
There are two types of mechanisms resulting in missing values in LC/MSMS experiments.
Missing values resulting from the absence of detection of a feature, despite ions being present at detectable concentrations. For example, this can happen in the case of ion suppression or as a result of the stochastic, data-dependent nature of the MS acquisition method. These missing values are expected to be randomly distributed in the data and are defined as missing at random (MAR) or missing completely at random (MCAR).
Biologically relevant missing values, resulting from the absence or low abundance of ions (below the limit of detection of the instrument). These missing values are not expected to be randomly distributed in the data and are defined as missing not at random (MNAR).
See Imputing quantitative proteomics data for more information.
First choose an imputation method (A):
knn: nearest neighbour averaging, as implemented in theimpute::impute.knnfunction. Note that this function is used with default parameters.MinDet: performs the imputation of left-censored missing data using a deterministic minimal value approach. Considering expression data withnsamples andpfeatures, for each sample, the missing entries are replaced with a minimal value observed in that sample. The minimal value observed is estimated as the q-th quantile (default q = 0.01) of the observed values in that sample. Implemented inimputeLCMD::impute.MinDet. Note that this function is used with default parameters.zero: replaces the missing values with 0.
Then click on Apply imputation (B). Once
the imputation has run, the page will display a post-imputation density
plot that you can colour by colData (C). Once the
imputation step is finished, do not forget to
Save the processed sets (D).
Aggregation
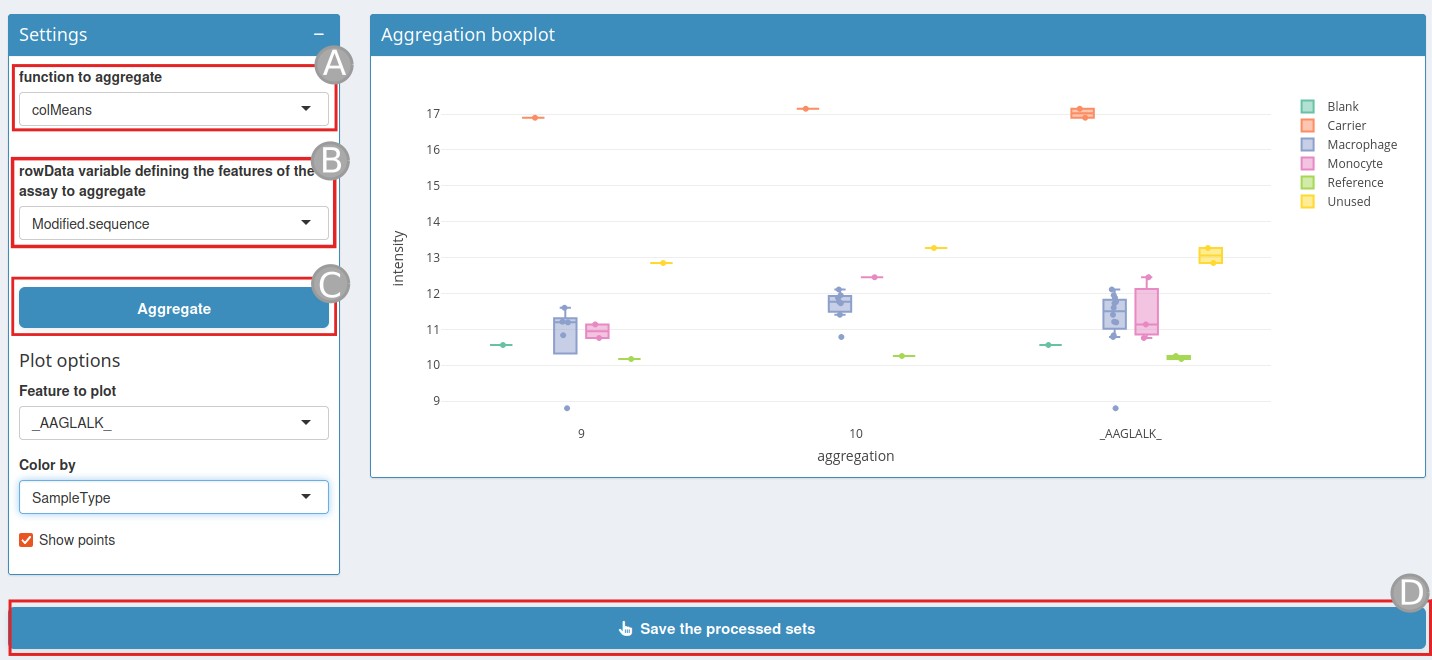
Aggregation page
At this stage, it is possible to use the peptide-level intensities or aggregate the peptide-level data into protein intensities.
To do this, a quantitative feature aggregation function is required.
This function, called Function to aggregate
(A), takes a matrix as input and returns a vector of length
equal to ncol(x). The function can be one of the following
types:
MsCoreUtils::medianPolish()fits an additive model (two-way decomposition) using Tukey’s median polish procedure withstats::medpolish().MsCoreUtils::robustSummarycalculates a robust aggregation usingMASS::rlm()(default).base::colMeans()to use the mean of each column.matrixStats::colMedians()to use the median of each column.base::colSums()to use the sum of each column.
See MsCoreUtils::aggregate_by_vector() for more
aggregation functions.
A rowData variable (B) is also needed to define how to
aggregate the features of the assays.
Once these two variables have been set, click on
Aggregate (C).
An aggregation boxplot will be displayed. This boxplot shows a
feature coloured by a colData value. Once the aggregation is done, click
on Save the processed sets.