Package: MsExperiment
Authors: Laurent Gatto [aut, cre] (ORCID: https://orcid.org/0000-0002-1520-2268), Johannes Rainer
[aut] (ORCID: https://orcid.org/0000-0002-6977-7147), Sebastian Gibb
[aut] (ORCID: https://orcid.org/0000-0001-7406-4443), Tuomas Borman
[ctb] (ORCID: https://orcid.org/0000-0002-8563-8884)
Last modified: 2026-02-06 13:11:59.632198
Compiled: Fri Feb 6 13:24:50 2026
Introduction
The goal of the MsExperiment
package is to provide a container for all data related to a mass
spectrometry (MS) experiment. Also other Bioconductor packages allow to
represent MS experiment data (such as the MSnbase
package). The MsExperiment however aims at being very
light-weight and flexible to accommodate all possible types of MS
experiments (proteomics, metabolomics, …) and all types of MS data
representations (chromatographic and spectral data, quantified features
etc). In addition, it allows to bundle additional files and data, such
as annotations, within the object.
In this vignette, we will describe how to create a
MsExperiment object and populate it with various types of
data.
We will also use the Spectra package to import MS data and thus load it here too.
Installation
The package can be installed with the BiocManager
package. To install BiocManager use
install.packages("BiocManager") and, after that,
BiocManager::install("MsExperiment") to install MsExperiment
which will install the package including all required dependencies.
Getting data
We will use a small subset of the PXD022816 project (Morgenstern et al. (2020)). The acquisitions correspond to a Pierce Thermo HeLa digestion standard, diluted to 50ng/uL with 97:3 + 0.1% formic acid, and acquired on a QExactive instrument.
Below, we use the rpx package
to access the project from the PRIDE repository, and download files of
interest. Note that these will automatically be cached in the
rpx packages’ cache directory.
## Loading PXD022816 from cache.
px## Project PXD022816 with 32 files
## ## Resource ID BFC1 in cache in /github/home/.cache/R/rpx.## [1] 'QEP2LC6_HeLa_50ng_251120_01-calib.mzID.gz' ... [32] 'checksum.txt'
## Use 'pxfiles(.)' to see all files.
pxfiles(px)## Project PXD022816 files (32):
## [remote] QEP2LC6_HeLa_50ng_251120_01-calib.mzID.gz
## [local] QEP2LC6_HeLa_50ng_251120_01-calib.mzML
## [remote] QEP2LC6_HeLa_50ng_251120_01.raw
## [remote] QEP2LC6_HeLa_50ng_251120_02-calib.mzID.gz
## [local] QEP2LC6_HeLa_50ng_251120_02-calib.mzML
## [remote] QEP2LC6_HeLa_50ng_251120_02.raw
## [remote] QEP2LC6_HeLa_50ng_251120_03-calib.mzID.gz
## [remote] QEP2LC6_HeLa_50ng_251120_03-calib.mzML
## [remote] QEP2LC6_HeLa_50ng_251120_03.raw
## [remote] QEP2LC6_HeLa_50ng_251120_04-calib.mzID.gz
## ...The project provides the vendor raw files, the converted mzML files as well as the identification mzid files. Let’s download fractions 1 and 2 of the mzML files.
If you run these commands interactively and it’s the first time you
use pxget(), you will be asked to create the
rpx cache directory - you can safelfy answer yes.
The files will then be downloaded. Next time you want to get the same
files, they will be loaded automatically from cache.
## Project PXD022816 files (32):
## [remote] QEP2LC6_HeLa_50ng_251120_01-calib.mzID.gz
## [local] QEP2LC6_HeLa_50ng_251120_01-calib.mzML
## [remote] QEP2LC6_HeLa_50ng_251120_01.raw
## [remote] QEP2LC6_HeLa_50ng_251120_02-calib.mzID.gz
## [local] QEP2LC6_HeLa_50ng_251120_02-calib.mzML
## [remote] QEP2LC6_HeLa_50ng_251120_02.raw
## [remote] QEP2LC6_HeLa_50ng_251120_03-calib.mzID.gz
## [remote] QEP2LC6_HeLa_50ng_251120_03-calib.mzML
## [remote] QEP2LC6_HeLa_50ng_251120_03.raw
## [remote] QEP2LC6_HeLa_50ng_251120_04-calib.mzID.gz
## ...## [1] "QEP2LC6_HeLa_50ng_251120_01-calib.mzML"
## [2] "QEP2LC6_HeLa_50ng_251120_02-calib.mzML"
fls <- pxget(px, i)## Loading QEP2LC6_HeLa_50ng_251120_01-calib.mzML from cache.## Loading QEP2LC6_HeLa_50ng_251120_02-calib.mzML from cache.
fls## [1] "/github/home/.cache/R/rpx/1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzML"
## [2] "/github/home/.cache/R/rpx/173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzML"Mass spectrometry experiment
Let’s start by creating an empty MsExperiment object
that we will populate with different pieces of data as we proceed with
the analysis of our data.
msexp <- MsExperiment()
msexp## Empty object of class MsExperimentExperiment files
Let’s now start with our MS experiment management by saving the
relevant files in a dedicated MsExperimentFiles object. In
addition to the mzML files, let’s also assume we have the human
proteomics fasta file ready. Later, when loading the raw data into R, we
will refer directly to the files in this MsExperimentFiles
object.
msfls <- MsExperimentFiles(mzmls = fls,
fasta = "homo_sapiens.fasta")
msfls## MsExperimentFiles of length 2
## [["mzmls"]] 1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzML ...
## [["fasta"]] homo_sapiens.fastaLet’s add these files to the main experiment management object:
experimentFiles(msexp) <- msfls
msexp## Object of class MsExperiment
## Files: mzmls, fastaExperimental design
The sampleData slot is used to describe the overall
experimental design of the experiment. It can be used to specify the
samples of the experiment and to relate them to the files that are part
of the experiment. There can be a one-to-one link between a sample and a
file, such as for example in label-free approaches, or one-to-many, in
labelled multiplexed approaches.
Here, we create a simple data frame with sample annotations that include the original file names and the respective fractions.
sampleData(msexp) <- DataFrame(
mzmls = basename(experimentFiles(msexp)[["mzmls"]]),
fractions = 1:2)
sampleData(msexp)## DataFrame with 2 rows and 2 columns
## mzmls fractions
## <character> <integer>
## 1 1739c1e6c3... 1
## 2 173927a78f... 2Raw data
We can now create a Spectra object containing the raw
data stored in the mzML files. If you are not familiar with the
Spectra object, please refer to the package
vignettes.
sp <- Spectra(experimentFiles(msexp)[["mzmls"]])
sp## MSn data (Spectra) with 58907 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 1 0.177987 1
## 2 1 0.599870 2
## 3 1 0.978849 3
## 4 1 1.363217 4
## 5 1 1.742965 5
## ... ... ... ...
## 58903 2 4198.59 29328
## 58904 1 4198.74 29329
## 58905 1 4199.11 29330
## 58906 1 4199.49 29331
## 58907 1 4199.87 29332
## ... 34 more variables/columns.
##
## file(s):
## 1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzML
## 173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzMLWe can now add this object to the main experiment management object:
spectra(msexp) <- sp
msexp## Object of class MsExperiment
## Files: mzmls, fasta
## Spectra: MS1 (12983) MS2 (45924)
## Experiment data: 2 sample(s)Third party applications
Let’s now assume we want to search the spectra in our mzML files
against the homo_sapiens.fasta file. To do so, we would
like to use a search engine such as MSGF+, that is run using the command
line and generates mzid files.
The command to run MSGF+ would look like this (see the manual page for details):
java -jar /path/to/MSGFPlus.jar \
-s input.mzML \
-o output.mzid
-d proteins.fasta \
-t 20ppm \ ## precursor mass tolerance
-tda 1 \ ## search decoy database
-m 0 \ ## fragmentation method as written in the spectrum or CID if no info
-int 1 ## Orbitrap/FTICR/LumosWe can easily build such a command for each of our input file:
mzids <- sub("mzML", "mzid", basename(experimentFiles(msexp)[["mzmls"]]))
paste0("java -jar /path/to/MSGFPlus.jar",
" -s ", experimentFiles(msexp)[["mzmls"]],
" -o ", mzids,
" -d ", experimentFiles(msexp)[["fasta"]],
" -t 20ppm",
" -m 0",
" int 1")## [1] "java -jar /path/to/MSGFPlus.jar -s /github/home/.cache/R/rpx/1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzML -o 1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzid -d homo_sapiens.fasta -t 20ppm -m 0 int 1"
## [2] "java -jar /path/to/MSGFPlus.jar -s /github/home/.cache/R/rpx/173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzML -o 173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzid -d homo_sapiens.fasta -t 20ppm -m 0 int 1"Here, for the sake of time and portability, we will not actually run MSGF+, but a simple shell script that will generate mzid files in a temporary R directory.
## [1] "/tmp/Rtmpen8GTM/1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzid"
## [2] "/tmp/Rtmpen8GTM/173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzid"
cmd <- paste("touch", output)
cmd## [1] "touch /tmp/Rtmpen8GTM/1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzid"
## [2] "touch /tmp/Rtmpen8GTM/173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzid"The cmd variable holds the two commands to be run on the
command line that will generate the new files. We can run each of these
commands with the system() function.
sapply(cmd, system)## touch /tmp/Rtmpen8GTM/1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzid
## 0
## touch /tmp/Rtmpen8GTM/173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzid
## 0Below, we add the names of the newly created files to our experiment:
experimentFiles(msexp)[["mzids"]] <- mzids
experimentFiles(msexp)## MsExperimentFiles of length 3
## [["mzmls"]] 1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzML ...
## [["fasta"]] homo_sapiens.fasta
## [["mzids"]] 1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzid ...
msexp## Object of class MsExperiment
## Files: mzmls, fasta, mzids
## Spectra: MS1 (12983) MS2 (45924)
## Experiment data: 2 sample(s)We can also decide to store the commands that were used to generate
the mzid files in the experiment’s metadata slot. Here, we use the
convention to name that metadata item "mzmls_to_mzids" to
document to input and output of these commands.
## $mzmls_to_mzids
## [1] "touch /tmp/Rtmpen8GTM/1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzid"
## [2] "touch /tmp/Rtmpen8GTM/173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzid"Finally, the existMsExperimentFiles() can be used at any
time to check which of files that are associated with an experiment
actually exist:
existMsExperimentFiles(msexp)## mzmls: 2 out of 2 exist(s)
## fasta: 0 out of 1 exist(s)
## mzids: 0 out of 2 exist(s)Saving and reusing experiments
The MsExperiment object has been used to store files and
data pertaining to a mass spectrometry experiment. It is now possible to
save that object and reload it later to recover all data and metadata.
See also section Using MsExperiment with
MsBackendSql below for an alternative to load/restore
defined MS experiments from an SQL database.
msexp <- readRDS("msexp.rds")
msexp## Object of class MsExperiment
## Files: mzmls, fasta, mzids
## Spectra: MS1 (12983) MS2 (45924)
## Experiment data: 2 sample(s)
experimentFiles(msexp)## MsExperimentFiles of length 3
## [["mzmls"]] 1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzML ...
## [["fasta"]] homo_sapiens.fasta
## [["mzids"]] 1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzid ...We can access the raw data as long as the mzML files that were used
to generate it still exist in their original location, which is the case
here as they were saved in the rpx cache directory.
sp <- spectra(msexp)
sp## MSn data (Spectra) with 58907 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 1 0.177987 1
## 2 1 0.599870 2
## 3 1 0.978849 3
## 4 1 1.363217 4
## 5 1 1.742965 5
## ... ... ... ...
## 58903 2 4198.59 29328
## 58904 1 4198.74 29329
## 58905 1 4199.11 29330
## 58906 1 4199.49 29331
## 58907 1 4199.87 29332
## ... 34 more variables/columns.
##
## file(s):
## 1739c1e6c38_QEP2LC6_HeLa_50ng_251120_01-calib.mzML
## 173927a78f33_QEP2LC6_HeLa_50ng_251120_02-calib.mzML
plotSpectra(sp[1000])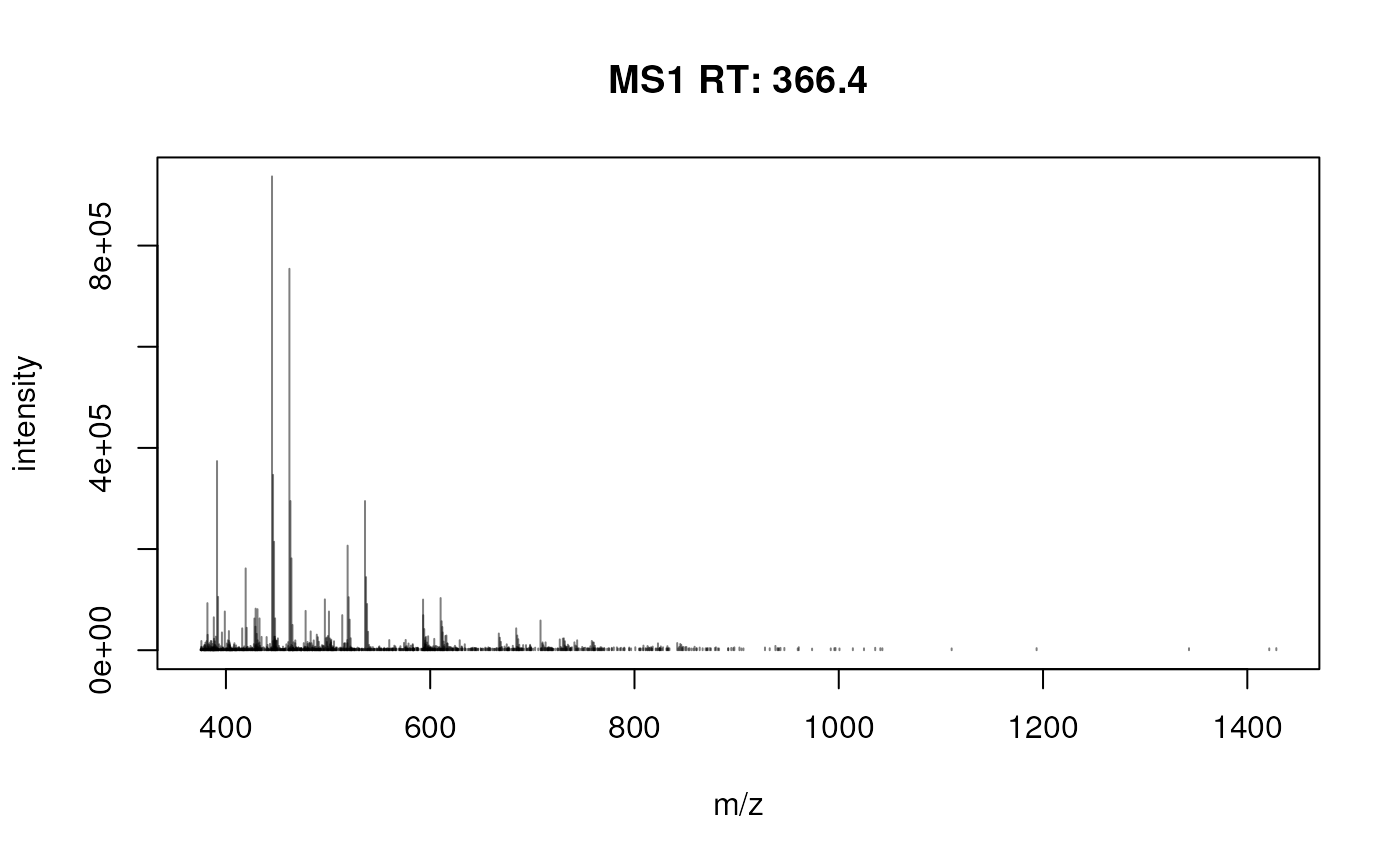
Linking experimental data to samples
For some experiments and data analyses an explicit link between data, data files and respective samples is required. Such links enable an easy (and error-free) subset or re-ordering of a whole experiment by sample and would also simplify coloring and labeling of the data depending on the sample or of its variables or conditions.
Below we generate an MsExperiment object for a simple
experiment consisting of a single sample measured in two different
injections to the same LC-MS setup.
lmse <- MsExperiment()
sd <- DataFrame(sample_id = c("QC1", "QC2"),
sample_name = c("QC Pool", "QC Pool"),
injection_idx = c(1, 3))
sampleData(lmse) <- sdWe next add mzML files to the experiment for the sample that was
measured. These are available through Bioconductor’s MsDataHub.
We add also an additional annotation file
"internal_standards.txt" to the experiment, which could be
e.g. a file with m/z and retention times of internal standards added to
the sample (note that such files don’t necessarily have to exist).
library(MsDataHub)
fls <- c(MsDataHub::X20171016_POOL_POS_1_105.134.mzML(),
MsDataHub::X20171016_POOL_POS_3_105.134.mzML())## see ?MsDataHub and browseVignettes('MsDataHub') for documentation## loading from cache## see ?MsDataHub and browseVignettes('MsDataHub') for documentation## loading from cache
basename(fls)## [1] "173941d49f52_7859" "17391674d18a_7860"
experimentFiles(lmse) <- MsExperimentFiles(
mzML_files = fls,
annotations = "internal_standards.txt")Next we load the MS data from the mzML files as a
Spectra object and add them to the experiment (see the
vignette of the Spectra
for details on import and representation of MS data).
sps <- Spectra(fls, backend = MsBackendMzR())
spectra(lmse) <- sps
lmse## Object of class MsExperiment
## Files: mzML_files, annotations
## Spectra: MS1 (1862)
## Experiment data: 2 sample(s)At this stage we have thus sample annotations and MS data in our
object, but no explicit relationships between them. Without such linking
between files and samples any subsetting would only subset the
sampleData but not any of the potentially associated files.
We next use the linkSpectraData() function to establish and
define such relationships. First we link the experimental files to the
samples: we want to link the first mzML file in the element called
"mzML_file" in the object’s experimentFiles to
the first row in sampleData and the second file to the
second row.
lmse <- linkSampleData(lmse, with = "experimentFiles.mzML_file",
sampleIndex = c(1, 2), withIndex = c(1, 2))To define the link we have thus to specify with which
element within our MsExperiment we want to link
samples. This can be done with the parameter with that
takes a single character representing the name
(address) of the data element. The name is a combination of the
name of the slot within the MsExperiment and the name of
the element (or column) within that slot separated by a
".". Using with = "experimentFiles.mzML_file"
means we want to link samples to values within the
"mzML_file" element of the object’s
experimentFiles slot - in other words, we want to link
samples to values in experimentFiles(lmse)$mzML_file. The
indices of the rows (samples) in sampleData and the indices
of the values in with to which we want to link the samples
can be defined with sampleIndex and withIndex.
In the example above we used sampleIndex = c(1, 2) and
withIndex = c(1, 2), thus, we want to link the first row in
sampleData to the first value in with and the
second row to the second value. See also the section Linking sample
data to other experimental data in the documentation of
MsExperiment for more information and details.
What happened internally by the call above is illustrated in the
figure below. The link is represented as a two-column integer
matrix with the indices of the linked sample in the first
and the indices of the associated elements in the second columns (this
matrix is essentially a cbind(sampleIndex, withIndex)).
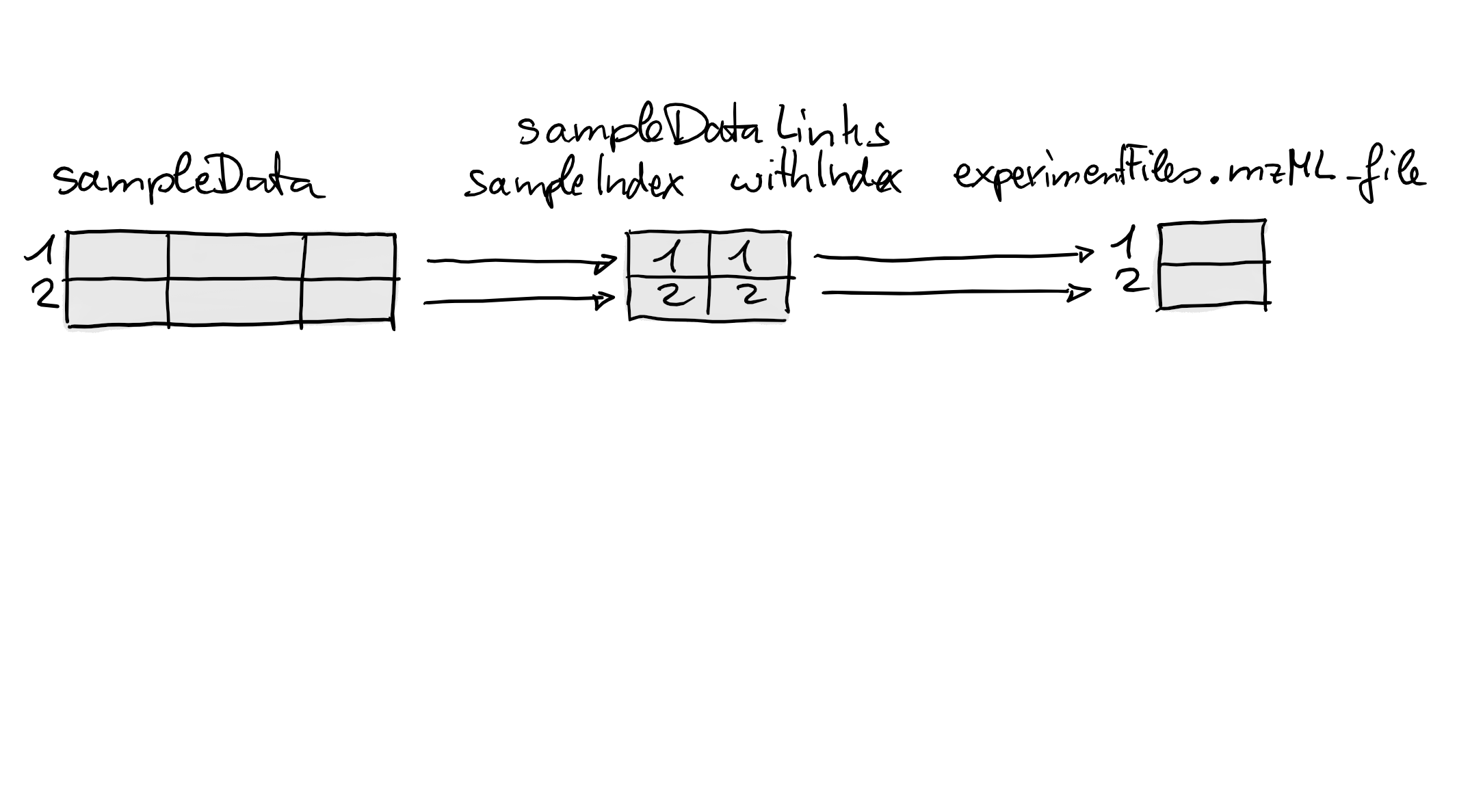
We next establish a second link between each sample and the
annotation file "internal_standards.txt" in
experimentFiles(lmse)$standards:
lmse <- linkSampleData(lmse, with = "experimentFiles.annotations",
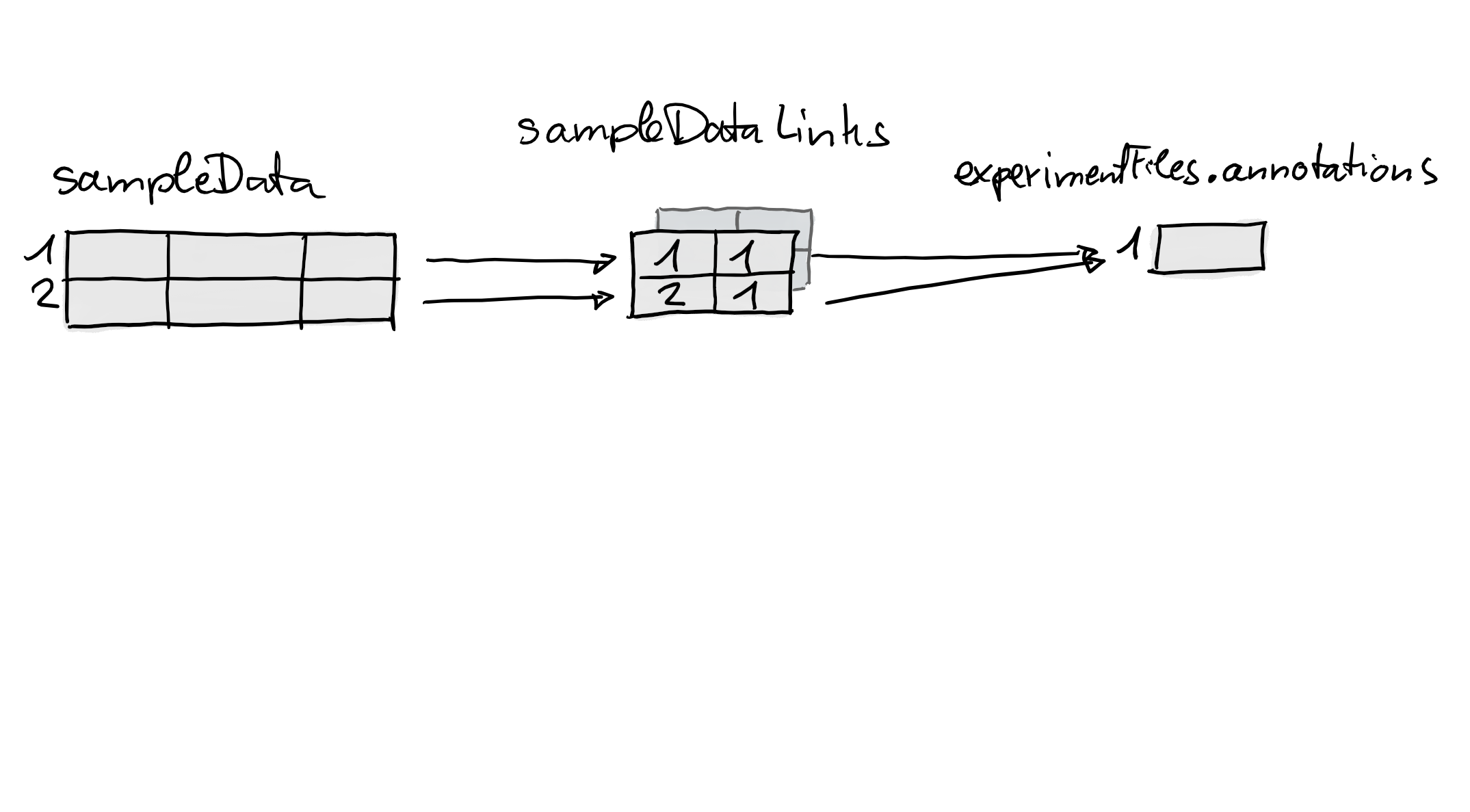
sampleIndex = c(1, 2), withIndex = c(1, 1))The figure below illustrates again what happened internally by this
call: a new link matrix was added establishing the relationship
between the two samples and the one value in
experimentFiles(lmse)$annotations.
It is thus also possible to link different samples to the same
element. We next link the spectra in the object to the individual
samples. We use for that an alternative way to specify the link without
the need to provide sampleIndex and withIndex.
Sample-to-data links can also be specified using a syntax similar to an
SQL join:
"sampleData.<column in sampleData> = <slot>.<element in slot>".
Links will be thus established between elements with matching values in
the specified data fields (i.e. between rows in sampleData
for which values in the specified column matches values in
<slot>.<element>). In order to use this
alternative approach to link spectra to the respective samples we have
to first add the (full) raw file name as an additional column to the
object’s sampleData. We can now add links between spectra
and samples by matching this raw file name to the original file name
from which the spectra were imported (which is available in the
"dataOrigin" spectra variable).
sampleData(lmse)$raw_file <- normalizePath(fls)
lmse <- linkSampleData(
lmse, with = "sampleData.raw_file = spectra.dataOrigin")The link was thus established between matching values in
sampleData(lmse)$raw_file and
spectra(lmse)$dataOrigin.
sampleData(lmse)$raw_file## [1] "/github/home/.cache/R/ExperimentHub/173941d49f52_7859"
## [2] "/github/home/.cache/R/ExperimentHub/17391674d18a_7860"## [1] "/github/home/.cache/R/ExperimentHub/173941d49f52_7859"
## [2] "/github/home/.cache/R/ExperimentHub/173941d49f52_7859"
## [3] "/github/home/.cache/R/ExperimentHub/173941d49f52_7859"
## [4] "/github/home/.cache/R/ExperimentHub/173941d49f52_7859"
## [5] "/github/home/.cache/R/ExperimentHub/173941d49f52_7859"
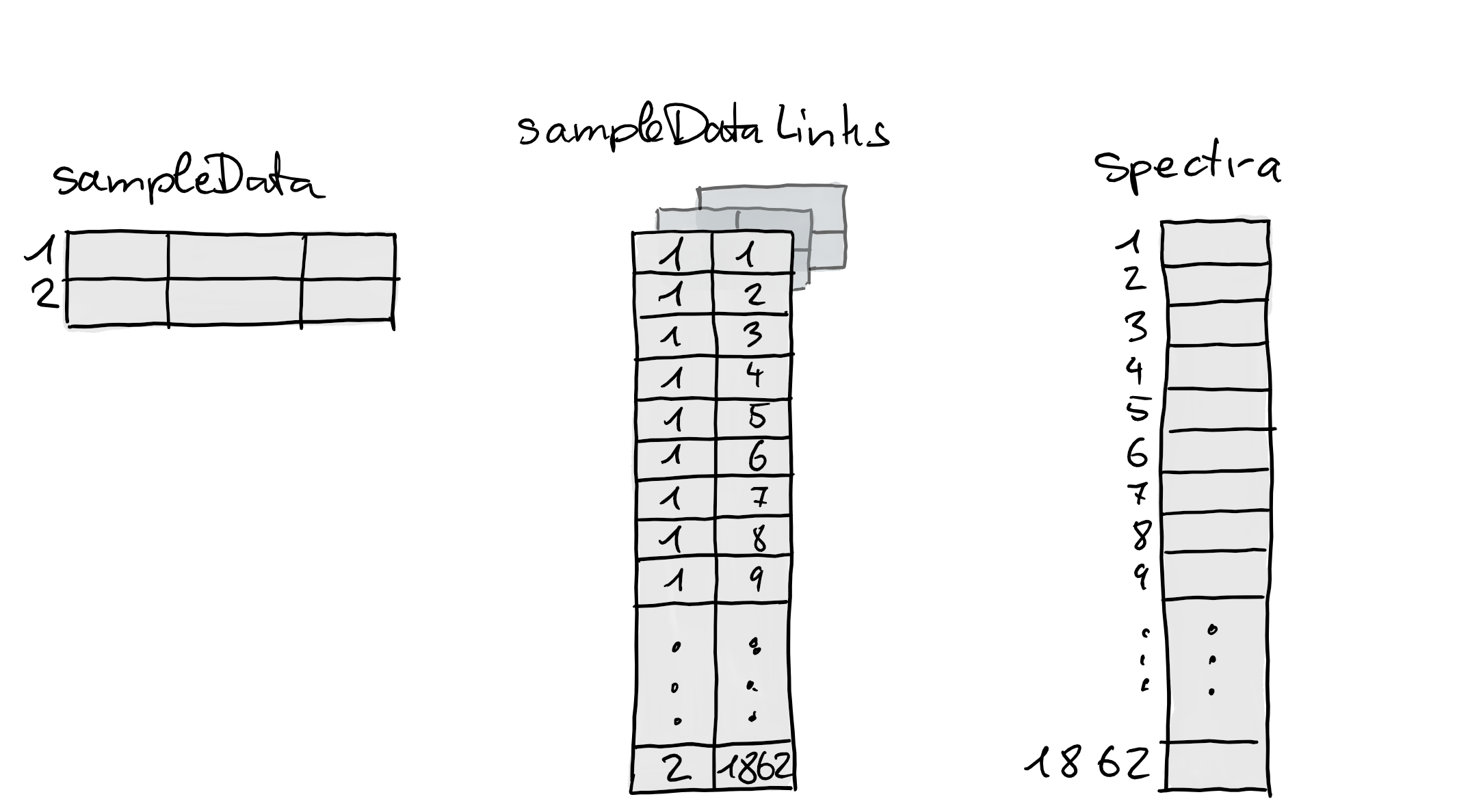
## [6] "/github/home/.cache/R/ExperimentHub/173941d49f52_7859"The figure below illustrates this link. With that last call we have
thus established links between samples and 3 different data elements in
the MsExperiment.
lmse## Object of class MsExperiment
## Files: mzML_files, annotations
## Spectra: MS1 (1862)
## Experiment data: 2 sample(s)
## Sample data links:
## - experimentFiles.mzML_file: 2 sample(s) to 2 element(s).
## - experimentFiles.annotations: 2 sample(s) to 1 element(s).
## - spectra: 2 sample(s) to 1862 element(s).A convenience function to quickly extract the index of a sample a
spectrum is associated with is spectraSampleIndex(). This
function returns an integer vector of length equal to the
number of spectra in the object with the row in the object’s
sampleData a spectrum is linked to, or
NA_integer_ if a spectrum is not linked to any sample.
#' Show the sample assignment for the first few spectra
spectraSampleIndex(lmse) |>
head()## [1] 1 1 1 1 1 1If we had also quantified feature values, we could also link
them to the samples. Below we create a simple, small
SummarizedExperiment to represent such quantified feature
values and add that to our experiment. To show that
MsExperiment supports also links between subsets of data
elements, we create a SummarizedExperiment that contains
values for an additional sample which is not present in our
sampleData. Also, we add samples in an arbitrary order.
library(SummarizedExperiment)
sd <- DataFrame(sample = c("QC2", "QC1", "QC3"), idx = c(3, 1, 5))
se <- SummarizedExperiment(colData = sd, assay = cbind(1:10, 11:20, 21:30))
qdata(lmse) <- seNext we link the samples in this SummarizedExperiment to
the samples to in the MsExperiment using matching values
between the "sample_id" column in the object’s
sampleData data frame and the column "sample"
in the SummarizedExperiment’s colData which is
stored in the @qdata slot. The naming convention to define
such columns is <slot name>.<column name>.
sampleData(lmse)$sample_id## [1] "QC1" "QC2"
qdata(lmse)$sample## [1] "QC2" "QC1" "QC3"
lmse <- linkSampleData(lmse, with = "sampleData.sample_id = qdata.sample")
lmse## Object of class MsExperiment
## Files: mzML_files, annotations
## Spectra: MS1 (1862)
## SummarizedExperiment: 10 feature(s)
## Experiment data: 2 sample(s)
## Sample data links:
## - experimentFiles.mzML_file: 2 sample(s) to 2 element(s).
## - experimentFiles.annotations: 2 sample(s) to 1 element(s).
## - spectra: 2 sample(s) to 1862 element(s).
## - qdata: 2 sample(s) to 2 column(s).The main advantage of all these links is that any subsetting of the experiment by sample will keep the (linked) data consistent. To illustrate this we subset below the experiment to the second sample.
b <- lmse[2]
b## Object of class MsExperiment
## Files: mzML_files, annotations, mzML_file
## Spectra: MS1 (931)
## SummarizedExperiment: 10 feature(s)
## Experiment data: 1 sample(s)
## Sample data links:
## - experimentFiles.mzML_file: 1 sample(s) to 1 element(s).
## - experimentFiles.annotations: 1 sample(s) to 1 element(s).
## - spectra: 1 sample(s) to 931 element(s).
## - qdata: 1 sample(s) to 1 column(s).The subset object contains now all data elements that are linked to
this second sample. Accessing the assay of the
SummarizedExperiment in qdata will thus return
only the quantified feature abundances for this second sample.
## [,1]
## [1,] 1
## [2,] 2
## [3,] 3
## [4,] 4
## [5,] 5
## [6,] 6
## [7,] 7
## [8,] 8
## [9,] 9
## [10,] 10But what happens for data elements that are not linked to any sample?
Below we add a data.frame as a metadata to the
experiment and subset the object again.
metadata(lmse)$other <- data.frame(
sample_name = c("study_1", "POOL", "study_2"),
index = 1:3)
b <- lmse[2]
metadata(b)## $other
## sample_name index
## 1 study_1 1
## 2 POOL 2
## 3 study_2 3By default, any element which is not linked to a sample is retained in the filtered/subset object.
We next link each sample to the second row in this data frame and subset the data again to the second sample.
lmse <- linkSampleData(lmse, with = "metadata.other",
sampleIndex = 1:2, withIndex = c(2, 2))
b <- lmse[2]
metadata(b)## $other
## sample_name index
## 2 POOL 2Subsetting thus retained only the row in the data frame for the linked sample. Obviously it is also possible to subset to multiple samples, in arbitrary order. Below we re-order our experiment.
lmse <- lmse[c(2, 1)]
sampleData(lmse)## DataFrame with 2 rows and 4 columns
## sample_id sample_name injection_idx raw_file
## <character> <character> <numeric> <character>
## 1 QC2 QC Pool 3 /github/ho...
## 2 QC1 QC Pool 1 /github/ho...The sample order is thus reversed and also all other linked elements
are re-ordered accordingly, such as "mzML_file" in the
object’s experimentFiles.
experimentFiles(lmse)$mzML_file## EH7810
## "/github/home/.cache/R/ExperimentHub/17391674d18a_7860"
## EH7809
## "/github/home/.cache/R/ExperimentHub/173941d49f52_7859"It is however important to note, that subsetting will also duplicate elements that are associated with multiple samples:
experimentFiles(lmse)$annotations## [1] "internal_standards.txt" "internal_standards.txt"Thus, while we added a single annotation file to the data
element "annotations" in experimentFiles,
after subsetting we ended up with two identical files. This duplication
of n:m relationships between samples to elements does however
not affect data consistency. A sample will always be linked to the
correct value/element.
Subset and filter MsExperiment
As already shown above, MsExperiment objects can be
subset with [ which will subset the data by sample.
Depending on whether relationships (links) between samples and any other
data within the object are present also these are correctly subset. In
addition to this general subset operation, it is possible to
individually filter the spectra data within an MsExperiment
using the filterSpectra function. This function takes any
filter function supported by Spectra with parameter
filter. Parameters for this filter function can be passed
through .... As an example we filter below the spectra data
of our MsExperiment keeping only spectra with an retention
time between 200 and 210 seconds.
#' Filter the Spectra using the `filterRt` function providing also the
#' parameters for this function.
res <- filterSpectra(lmse, filterRt, rt = c(200, 210))
res## Object of class MsExperiment
## Files: mzML_files, annotations, mzML_file
## Spectra: MS1 (72)
## SummarizedExperiment: 10 feature(s)
## Experiment data: 2 sample(s)
## Sample data links:
## - experimentFiles.mzML_file: 2 sample(s) to 2 element(s).
## - experimentFiles.annotations: 2 sample(s) to 2 element(s).
## - spectra: 2 sample(s) to 72 element(s).
## - qdata: 2 sample(s) to 2 column(s).
## - metadata.other: 2 sample(s) to 2 element(s).The resulting MsExperiment contains now much fewer
spectra. filterSpectra did only filter the spectra data,
but not any of the other data slots. It did however update and
consolidate the relationships between samples and spectra (if present)
after filtering:
#' Extract spectra of the second sample after filtering
spectra(res[2L])## MSn data (Spectra) with 36 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 1 200.049 717
## 2 1 200.328 718
## 3 1 200.607 719
## 4 1 200.886 720
## 5 1 201.165 721
## ... ... ... ...
## 32 1 208.698 748
## 33 1 208.977 749
## 34 1 209.256 750
## 35 1 209.535 751
## 36 1 209.814 752
## ... 34 more variables/columns.
##
## file(s):
## 173941d49f52_7859
## Processing:
## Filter: select retention time [200..210] on MS level(s) [Fri Feb 6 13:25:12 2026]Using MsExperiment with MsBackendSql
The MsBackendSql
provides functionality to store mass spectrometry data in a SQL database
and to represent such data as a Spectra object
(through its MsBackendSql or
MsBackendOfflineSql backends). Next to the MS data, it is
also possible to store sample data in such SQL databases hence allowing
to create self-contained databases for MS experiments. This simplifies
the process to load pre-defined MS experiment or to share experiment
data across analyses or with a collaborator. In this section we show how
MS data and sample information from an experiment can be stored into a
SQL database and how that data can then be loaded again as a
MsExperiment.
Below we first define the raw data files of the experiment. In our example we use two test files provided by Bioconductor’s MsDataHub.
fls <- c(MsDataHub::X20171016_POOL_POS_1_105.134.mzML(),
MsDataHub::X20171016_POOL_POS_3_105.134.mzML())## see ?MsDataHub and browseVignettes('MsDataHub') for documentation## loading from cache## see ?MsDataHub and browseVignettes('MsDataHub') for documentation## loading from cacheWe can then use the createMsBackendSqlDatabase function
from the MsBackendSql
package to write the data into a SQLite database. SQLite databases are
particularly easy to create, use and also to share, but for large
experiments it is suggested to use more powerful database engines, such
as for example MariaDB (see also the documentation and vignette
of the MsBackendSql
package for parameters and settings for such large databases).
Also, in our example we store the SQLite database into a data temporary file, but for a real use case we would want to use a real file.
library(MsBackendSql)
library(RSQLite)
#' Create the SQLite database where to store the data. For our
#' example we create the database in a temporary file.
sql_file <- tempfile()
con <- dbConnect(SQLite(), dbname = sql_file)
#' Write the MS data to the.
createMsBackendSqlDatabase(dbcon = con, fls)## Importing data ...## [==========================================================] 1/1 (100%) in 3s
## Creating indices .... Done## [1] TRUEWe can now create a Spectra object representing the MS
data in that database using either a MsBackendSql or
MsBackendOfflineSql backend.
#' Load the database as a Spectra object
sps <- Spectra(sql_file, source = MsBackendOfflineSql(), drv = SQLite())
sps## MSn data (Spectra) with 1862 spectra in a MsBackendOfflineSql backend:
## msLevel precursorMz polarity
## <integer> <numeric> <integer>
## 1 1 NA 1
## 2 1 NA 1
## 3 1 NA 1
## 4 1 NA 1
## 5 1 NA 1
## ... ... ... ...
## 1858 1 NA 1
## 1859 1 NA 1
## 1860 1 NA 1
## 1861 1 NA 1
## 1862 1 NA 1
## ... 35 more variables/columns.
## Use 'spectraVariables' to list all of them.
## Database: /tmp/Rtmpen8GTM/file1e6952769a2Next we define sample annotations for the two files and create a
MsExperiment with the MS data and the related sample
annotations.
#' Define sample annotations. Ideally all experiment-relavant information
#' should be specified.
sdata <- data.frame(file_name = basename(fls),
sample_name = c("QC_POOL_1", "QC_POOL_3"),
sample_group = c("QC", "QC"))
#' To simplify subsequent linking between samples and data files we
#' define a new spectra variable with the file names of the raw data files.
sps$file_name <- basename(dataOrigin(sps))
#' Create the MsExperiment for that data
mse <- MsExperiment(sampleData = sdata, spectra = sps)Finally we link the MS spectra to the individual samples using the file names of the original data files.
#' Establish the mapping between spectra and samples
mse <- linkSampleData(mse, with = "sampleData.file_name = spectra.file_name")
mse## Object of class MsExperiment
## Spectra: MS1 (1862)
## Experiment data: 2 sample(s)
## Sample data links:
## - spectra: 2 sample(s) to 1862 element(s).With sample annotations and the mapping between samples and spectra
defined, we can next write this information to the SQL database
containing already the MS data of our experiment: the
dbWriteSampleData function writes, for
MsExperiment objects that use a Spectra with a
MsBackendSql (or MsBackendOfflineSql) backend
the available sample data to additional database tables in the
database.
#' Write sample data (and mapping between samples and spectra) to the
#' database
dbWriteSampleData(mse)The SQL database does now contain the MS data, the sample annotations and the mapping between samples and spectra. Creation of such a database needs to be performed only once for an experiment. The associated SQLite database file could now be shared with a collaborator or used across different analyses without the need to share or copy also the raw data files or sample annotations (which would usually needed to be provided as a separate file).
To load the MS experiment data again into R, we first need to create
a Spectra object with a MsBackendSql or
MsBackendOfflineSql backend for that SQL database.
#' Create a Spectra object for the MS data of that database
sps <- Spectra(sql_file, source = MsBackendOfflineSql(), drv = SQLite())We then pass this Spectra object with parameter
spectra to the MsExperiment constructor call.
The function will then read also sample data from the database (if
available) as well as the mapping between samples and spectra to restore
our MS experiment.
#' Create the MsExperiment reading MS data and sample data from
#' the database
mse <- MsExperiment(spectra = sps)
mse## Object of class MsExperiment
## Spectra: MS1 (1862)
## Experiment data: 2 sample(s)
## Sample data links:
## - spectra: 2 sample(s) to 1862 element(s).We have thus now the full MS experiment data available.
For smaller experiments, such SQL databases could also be created
with an alternative, eventually simpler, approach. For that we first use
the readMsExperiment function to create an
MsExperiment from the file names of the raw MS data files
and a data.frame with the associated sample annotations. We
re-use here the variables fls with the data file names and
sdata with the sample data.
#' Create an MsExperiment with data from two (raw) data files
#' and associated sample annotations.
mse <- readMsExperiment(spectraFiles = fls, sampleData = sdata)
mse## Object of class MsExperiment
## Spectra: MS1 (1862)
## Experiment data: 2 sample(s)
## Sample data links:
## - spectra: 2 sample(s) to 1862 element(s).The MS data in that MsExperiment is represented by a
Spectra object using the MsBackendMzR:
#' Show the Spectra from the experiment
spectra(mse)## MSn data (Spectra) with 1862 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 1 0.280 1
## 2 1 0.559 2
## 3 1 0.838 3
## 4 1 1.117 4
## 5 1 1.396 5
## ... ... ... ...
## 1858 1 258.636 927
## 1859 1 258.915 928
## 1860 1 259.194 929
## 1861 1 259.473 930
## 1862 1 259.752 931
## ... 34 more variables/columns.
##
## file(s):
## 173941d49f52_7859
## 17391674d18a_7860We next change the backend of this Spectra object using
the setBackend function to a
MsBackendOfflineSql. We choose again a SQLite database
format and store the data to a temporary file.
#' Define the SQLite database file
sql_file2 <- tempfile()
#' Change the backend of the Spectra data
spectra(mse) <- setBackend(spectra(mse), MsBackendOfflineSql(),
dbname = sql_file2, drv = SQLite())## Importing data ...## [============================>-----------------------------] 1/2 ( 50%) in 0s[==========================================================] 2/2 (100%) in 1s
## Creating indices .... Done
spectra(mse)## MSn data (Spectra) with 1862 spectra in a MsBackendOfflineSql backend:
## msLevel precursorMz polarity
## <integer> <numeric> <integer>
## 1 1 NA 1
## 2 1 NA 1
## 3 1 NA 1
## 4 1 NA 1
## 5 1 NA 1
## ... ... ... ...
## 1858 1 NA 1
## 1859 1 NA 1
## 1860 1 NA 1
## 1861 1 NA 1
## 1862 1 NA 1
## ... 35 more variables/columns.
## Use 'spectraVariables' to list all of them.
## Database: /tmp/Rtmpen8GTM/file1e6956f6775b
## Processing:
## Switch backend from MsBackendMzR to MsBackendOfflineSql [Fri Feb 6 13:25:18 2026]All MS data is now stored in the SQLite database and the
Spectra object of our experiment uses the
MsBackendOfflineSql to represent/interface this data. To
store also the sample data to the database we can use the
dbWriteSampleData as before.
#' Store also sample data to the database
dbWriteSampleData(mse)In addition to create self-contained SQL databases for an experiment, this code could also be used within an analysis workflow to serialize/de-serialize results, always with the advantage of keeping the full experiment data in a single place (file) in a language-agnostic format.
We can for example use basic functions from the DBI/RSQLite packages (or simple SQL commands) to access the data in the database. Below we connect to the database and list the available tables.
#' Connect to the database
con <- dbConnect(SQLite(), dbname = sql_file2)
#' List the available database tables
dbListTables(con)## [1] "msms_spectrum" "msms_spectrum_peak_blob2"
## [3] "sample_data" "sample_to_msms_spectrum"The msms_spectrum table contains general metadata for the individual MS spectra, msms_spectrum_peak_blob the actual peak data (m/z and intensity values), sample_data the sample annotations and sample_to_msms_spectrum the link between samples and spectra. Below we retrieve the content of the sample_data table.
#' Get the content of the sample_data table
dbGetQuery(con, "select * from sample_data")## file_name sample_name sample_group
## 1 173941d49f52_7859 QC_POOL_1 QC
## 2 17391674d18a_7860 QC_POOL_3 QC
## spectraOrigin sample_id_
## 1 /github/home/.cache/R/ExperimentHub/173941d49f52_7859 1
## 2 /github/home/.cache/R/ExperimentHub/17391674d18a_7860 2
#' Close the connection to the database
dbDisconnect(con)Session information
## R Under development (unstable) (2026-02-01 r89366)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] RSQLite_2.4.6 MsBackendSql_1.11.2
## [3] SummarizedExperiment_1.41.0 Biobase_2.71.0
## [5] GenomicRanges_1.63.1 Seqinfo_1.1.0
## [7] IRanges_2.45.0 MatrixGenerics_1.23.0
## [9] matrixStats_1.5.0 MsDataHub_1.11.0
## [11] rpx_2.15.1 Spectra_1.21.1
## [13] BiocParallel_1.45.0 S4Vectors_0.49.0
## [15] BiocGenerics_0.57.0 generics_0.1.4
## [17] MsExperiment_1.13.1 ProtGenerics_1.39.2
## [19] BiocStyle_2.39.0
##
## loaded via a namespace (and not attached):
## [1] DBI_1.2.3 bitops_1.0-9
## [3] httr2_1.2.2 rlang_1.1.7
## [5] magrittr_2.0.4 clue_0.3-66
## [7] otel_0.2.0 compiler_4.6.0
## [9] png_0.1-8 systemfonts_1.3.1
## [11] vctrs_0.7.1 reshape2_1.4.5
## [13] stringr_1.6.0 crayon_1.5.3
## [15] pkgconfig_2.0.3 MetaboCoreUtils_1.19.1
## [17] fastmap_1.2.0 dbplyr_2.5.1
## [19] XVector_0.51.0 rmarkdown_2.30
## [21] ragg_1.5.0 purrr_1.2.1
## [23] bit_4.6.0 xfun_0.56
## [25] MultiAssayExperiment_1.37.2 cachem_1.1.0
## [27] jsonlite_2.0.0 progress_1.2.3
## [29] blob_1.3.0 DelayedArray_0.37.0
## [31] prettyunits_1.2.0 parallel_4.6.0
## [33] cluster_2.1.8.2 R6_2.6.1
## [35] bslib_0.10.0 stringi_1.8.7
## [37] jquerylib_0.1.4 Rcpp_1.1.1
## [39] bookdown_0.46 knitr_1.51
## [41] Matrix_1.7-4 igraph_2.2.1
## [43] tidyselect_1.2.1 abind_1.4-8
## [45] yaml_2.3.12 codetools_0.2-20
## [47] curl_7.0.0 lattice_0.22-7
## [49] tibble_3.3.1 plyr_1.8.9
## [51] withr_3.0.2 KEGGREST_1.51.1
## [53] evaluate_1.0.5 desc_1.4.3
## [55] BiocFileCache_3.1.0 xml2_1.5.2
## [57] Biostrings_2.79.4 ExperimentHub_3.1.0
## [59] pillar_1.11.1 BiocManager_1.30.27
## [61] filelock_1.0.3 ncdf4_1.24
## [63] RCurl_1.98-1.17 hms_1.1.4
## [65] BiocVersion_3.23.1 glue_1.8.0
## [67] lazyeval_0.2.2 tools_4.6.0
## [69] AnnotationHub_4.1.0 data.table_1.18.2.1
## [71] QFeatures_1.21.0 mzR_2.45.0
## [73] fs_1.6.6 fastmatch_1.1-8
## [75] grid_4.6.0 tidyr_1.3.2
## [77] MsCoreUtils_1.23.2 AnnotationDbi_1.73.0
## [79] cli_3.6.5 rappdirs_0.3.4
## [81] textshaping_1.0.4 S4Arrays_1.11.1
## [83] dplyr_1.2.0 AnnotationFilter_1.35.0
## [85] sass_0.4.10 digest_0.6.39
## [87] SparseArray_1.11.10 htmlwidgets_1.6.4
## [89] memoise_2.0.1 htmltools_0.5.9
## [91] pkgdown_2.2.0.9000 lifecycle_1.0.5
## [93] httr_1.4.7 bit64_4.6.0-1
## [95] MASS_7.3-65