Package: Spectra
Authors: RforMassSpectrometry Package Maintainer [cre],
Laurent Gatto [aut] (ORCID: https://orcid.org/0000-0002-1520-2268), Johannes Rainer
[aut] (ORCID: https://orcid.org/0000-0002-6977-7147), Sebastian Gibb
[aut] (ORCID: https://orcid.org/0000-0001-7406-4443), Philippine
Louail [aut] (ORCID: https://orcid.org/0009-0007-5429-6846), Jan Stanstrup
[ctb] (ORCID: https://orcid.org/0000-0003-0541-7369), Nir Shahaf
[ctb], Mar Garcia-Aloy [ctb] (ORCID: https://orcid.org/0000-0002-1330-6610), Guillaume
Deflandre [ctb] (ORCID: https://orcid.org/0009-0008-1257-2416), Ahlam Mentag
[ctb] (ORCID: https://orcid.org/0009-0008-5438-7067)
Last modified: 2026-06-12 14:09:35.718635
Compiled: Fri Jun 12 14:35:35 2026
Introduction
The Spectra
package provides a scalable and flexible infrastructure to represent,
retrieve and handle mass spectrometry (MS) data. The
Spectra object provides the user with a single standardized
interface to access and manipulate MS data while supporting, through the
concept of exchangeable backends, a large variety of different
ways to store and retrieve mass spectrometry data. Such backends range
from mzML/mzXML/CDF files, simple flat files, or database systems.
This vignette provides general examples and descriptions for the Spectra package. Additional information and tutorials are available, such as SpectraTutorials, MetaboAnnotationTutorials, or also in (Rainer et al. 2022). For information on how to handle and (parallel) process large-scale data sets see the Large-scale data handling and processing with Spectra vignette.
Installation
The package can be installed with the BiocManager package.
To install BiocManager use
install.packages("BiocManager") and, after that,
BiocManager::install("Spectra") to install
Spectra.
General usage
Mass spectrometry data in Spectra objects can be thought
of as a list of individual spectra, with each spectrum having a set of
variables associated with it. Besides core spectra variables
(such as MS level or retention time) an arbitrary number of optional
variables can be assigned to a spectrum. The core spectra variables all
have their own accessor method and it is guaranteed that a value is
returned by it (or NA if the information is not available).
The core variables and their data type are (alphabetically ordered):
-
acquisitionNum
integer(1): the index of acquisition of a spectrum during a MS run. -
centroided
logical(1): whether the spectrum is in profile or centroid mode. -
collisionEnergy
numeric(1): collision energy used to create an MSn spectrum. -
dataOrigin
character(1): the origin of the spectrum’s data, e.g. the mzML file from which it was read. -
dataStorage
character(1): the (current) storage location of the spectrum data. This value depends on the backend used to handle and provide the data. For an in-memory backend like theMsBackendDataFramethis will be"<memory>", for an on-disk backend such as theMsBackendHdf5Peaksit will be the name of the HDF5 file where the spectrum’s peak data is stored. -
intensity
numeric: intensity values for the spectrum’s peaks. -
isolationWindowLowerMz
numeric(1): lower m/z for the isolation window in which the (MSn) spectrum was measured. -
isolationWindowTargetMz
numeric(1): the target m/z for the isolation window in which the (MSn) spectrum was measured. -
isolationWindowUpperMz
numeric(1): upper m/z for the isolation window in which the (MSn) spectrum was measured. -
msLevel
integer(1): the MS level of the spectrum. -
mz
numeric: the m/z values for the spectrum’s peaks. -
polarity
integer(1): the polarity of the spectrum (0and1representing negative and positive polarity, respectively). -
precScanNum
integer(1): the scan (acquisition) number of the precursor for an MSn spectrum. -
precursorCharge
integer(1): the charge of the precursor of an MSn spectrum. -
precursorIntensity
numeric(1): the intensity of the precursor of an MSn spectrum. -
precursorMz
numeric(1): the m/z of the precursor of an MSn spectrum. -
rtime
numeric(1): the retention time of a spectrum. -
scanIndex
integer(1): the index of a spectrum within a (raw) file. -
smoothed
logical(1): whether the spectrum was smoothed.
For details on the individual variables and their getter/setter
function see the help for Spectra (?Spectra).
Also note that these variables are suggested, but not required to
characterize a spectrum. Also, some only make sense for MSn, but not for
MS1 spectra.
Creating Spectra objects
The simplest way to create a Spectra object is by
defining a DataFrame with the corresponding spectra data
(using the corresponding spectra variable names as column names) and
passing that to the Spectra constructor function. Below we
create such an object for a set of 3 spectra providing their MS level,
olarity but also additional annotations such as their ID in HMDB (human metabolome database) and their
name. The m/z and intensity values for each spectrum have to be provided
as a list of numeric values.
library(Spectra)
spd <- DataFrame(
msLevel = c(2L, 2L, 2L),
polarity = c(1L, 1L, 1L),
id = c("HMDB0000001", "HMDB0000001", "HMDB0001847"),
name = c("1-Methylhistidine", "1-Methylhistidine", "Caffeine"))
## Assign m/z and intensity values.
spd$mz <- list(
c(109.2, 124.2, 124.5, 170.16, 170.52),
c(83.1, 96.12, 97.14, 109.14, 124.08, 125.1, 170.16),
c(56.0494, 69.0447, 83.0603, 109.0395, 110.0712,
111.0551, 123.0429, 138.0662, 195.0876))
spd$intensity <- list(
c(3.407, 47.494, 3.094, 100.0, 13.240),
c(6.685, 4.381, 3.022, 16.708, 100.0, 4.565, 40.643),
c(0.459, 2.585, 2.446, 0.508, 8.968, 0.524, 0.974, 100.0, 40.994))
sps <- Spectra(spd)
sps## MSn data (Spectra) with 3 spectra in a MsBackendMemory backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 NA NA
## 2 2 NA NA
## 3 2 NA NA
## ... 18 more variables/columns.Alternatively, it is possible to import spectra data from mass
spectrometry raw files in mzML/mzXML or CDF format. Below we create a
Spectra object from two mzML files and define to use a
MsBackendMzR backend to store the data (note that
this requires the mzR package
to be installed). This backend, specifically designed for raw MS data,
keeps only a subset of spectra variables in memory while reading the m/z
and intensity values from the original data files only on demand. See
section Backends for more details on backends
and their properties.
register(SerialParam())
#' define the file names of the data to import; data provided through
#' the MsDataHub package
fls <- c(MsDataHub::X20171016_POOL_POS_1_105.134.mzML(),
MsDataHub::X20171016_POOL_POS_3_105.134.mzML())
sps_sciex <- Spectra(fls, source = MsBackendMzR())
sps_sciex## MSn data (Spectra) with 1862 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 1 0.280 1
## 2 1 0.559 2
## 3 1 0.838 3
## 4 1 1.117 4
## 5 1 1.396 5
## ... ... ... ...
## 1858 1 258.636 927
## 1859 1 258.915 928
## 1860 1 259.194 929
## 1861 1 259.473 930
## 1862 1 259.752 931
## ... 34 more variables/columns.
##
## file(s):
## 7482fe40e87_7859
## 74890c8574_7860Note that we disabled any parallel processing using the
register(SerialParam()) call. Many functions for
Spectra objects support parallel processing and will by
default use the globally defined setup. On Windows machines only
BiocParallel::SnowParam() is supported, which however will
start a new R process for each parallel process for any operation -
which can take up to 30-40 seconds. Hence, for Windows, it is suggested
to either register and initiate the parallel processing system globally
(using e.g. register(bppstart(SnowParam(4))) to start a
parallel processing setup with 4 processes), or to disable it (using
register(SerialParam())).
The Spectra object sps_sciex allows now to
access spectra data from 1862 MS1 spectra and uses
MsBackendMzR as backend (the Spectra object
sps created in the previous code block uses the default
MsBackendMemory).
Accessing spectrum data
As detailed above Spectra objects can contain an
arbitrary number of properties of a spectrum (so called spectra
variables). The available variables can be listed with the
spectraVariables() method:
spectraVariables(sps)## [1] "msLevel" "rtime"
## [3] "acquisitionNum" "scanIndex"
## [5] "dataStorage" "dataOrigin"
## [7] "centroided" "smoothed"
## [9] "polarity" "precScanNum"
## [11] "precursorMz" "precursorIntensity"
## [13] "precursorCharge" "collisionEnergy"
## [15] "isolationWindowLowerMz" "isolationWindowTargetMz"
## [17] "isolationWindowUpperMz" "id"
## [19] "name"
spectraVariables(sps_sciex)## [1] "msLevel" "rtime"
## [3] "acquisitionNum" "scanIndex"
## [5] "dataStorage" "dataOrigin"
## [7] "centroided" "smoothed"
## [9] "polarity" "precScanNum"
## [11] "precursorMz" "precursorIntensity"
## [13] "precursorCharge" "collisionEnergy"
## [15] "isolationWindowLowerMz" "isolationWindowTargetMz"
## [17] "isolationWindowUpperMz" "peaksCount"
## [19] "totIonCurrent" "basePeakMZ"
## [21] "basePeakIntensity" "electronBeamEnergy"
## [23] "ionisationEnergy" "lowMZ"
## [25] "highMZ" "mergedScan"
## [27] "mergedResultScanNum" "mergedResultStartScanNum"
## [29] "mergedResultEndScanNum" "injectionTime"
## [31] "filterString" "spectrumId"
## [33] "ionMobilityDriftTime" "scanWindowLowerLimit"
## [35] "scanWindowUpperLimit"The two Spectra contain a different set of variables:
besides "msLevel", "polarity",
"id" and "name", that were specified for the
Spectra object sps, it contains more variables
such as "rtime", "acquisitionNum" and
"scanIndex". These are part of the core variables
defining a spectrum and for all of these accessor methods exist. Below
we use msLevel() and rtime() to access the MS
levels and retention times for the spectra in sps.
msLevel(sps)## [1] 2 2 2
rtime(sps)## [1] NA NA NAWe did not specify retention times for the spectra in
sps thus NA is returned for them. The
Spectra object sps_sciex contains many more
variables, all of which were extracted from the mzML files. Below we
extract the retention times for the first spectra in the object.
## [1] 0.280 0.559 0.838 1.117 1.396 1.675Note that in addition to the accessor functions it is also possible
to use $ to extract a specific spectra variable. To extract
the name of the compounds in sps we can use
sps$name, or, to extract the MS levels
sps$msLevel.
sps$name## [1] "1-Methylhistidine" "1-Methylhistidine" "Caffeine"
sps$msLevel## [1] 2 2 2We could also replace specific spectra variables using either the
dedicated method or $. Below we specify that all spectra in
sps represent centroided data.
sps$centroided <- TRUE
centroided(sps)## [1] TRUE TRUE TRUEThe $ operator can also be used to add arbitrary new
spectra variables to a Spectra object. Below we add the
SPLASH key to each of the spectra.
sps$splash <- c(
"splash10-00di-0900000000-037d24a7d65676b7e356",
"splash10-00di-0900000000-03e99316bd6c098f5d11",
"splash10-000i-0900000000-9af60e39c843cb715435")This new spectra variable will now be listed as an additional
variable in the result of the spectraVariables() function
and we can directly access its content with sps$splash.
Each spectrum can have a different number of mass peaks, each
consisting of a mass-to-charge (m/z) and associated intensity value.
These can be extracted with the mz() or
intensity() functions, each of which return a
list of numeric values.
mz(sps)## NumericList of length 3
## [[1]] 109.2 124.2 124.5 170.16 170.52
## [[2]] 83.1 96.12 97.14 109.14 124.08 125.1 170.16
## [[3]] 56.0494 69.0447 83.0603 109.0395 110.0712 111.0551 123.0429 138.0662 195.0876
intensity(sps)## NumericList of length 3
## [[1]] 3.407 47.494 3.094 100 13.24
## [[2]] 6.685 4.381 3.022 16.708 100 4.565 40.643
## [[3]] 0.459 2.585 2.446 0.508 8.968 0.524 0.974 100 40.994Peak data can also be extracted with the peaksData()
function that returns a list of numerical matrices with peak
variables such as m/z and intensity values. Which peak variables
are available in a Spectra object can be determined with
the peaksVariables() function.
peaksVariables(sps)## [1] "mz" "intensity"These can be passed to the peaksData() function with
parameter columns to extract the peak variables of
interest. By default peaksData() extracts m/z and intensity
values.
pks <- peaksData(sps)
pks[[1]]## mz intensity
## [1,] 109.20 3.407
## [2,] 124.20 47.494
## [3,] 124.50 3.094
## [4,] 170.16 100.000
## [5,] 170.52 13.240Note that we would get the same result by using the as()
method to coerce a Spectra object to a list or
SimpleList:
as(sps, "SimpleList")## List of length 3The spectraData() function returns a
DataFrame with the full data for each spectrum (except m/z
and intensity values), or with selected spectra variables (which can be
specified with the columns parameter). Below we extract the
spectra data for variables "msLevel", "id" and
"name".
spectraData(sps, columns = c("msLevel", "id", "name"))## DataFrame with 3 rows and 3 columns
## msLevel id name
## <integer> <character> <character>
## 1 2 HMDB0000001 1-Methylhistidine
## 2 2 HMDB0000001 1-Methylhistidine
## 3 2 HMDB0001847 CaffeineSpectra are one-dimensional objects storing spectra,
even from different files or samples, in a single list. Specific
variables have thus to be used to define the originating file from which
they were extracted or the sample in which they were measured. The
data origin of each spectrum can be extracted with the
dataOrigin() function. For sps, the
Spectra created from a DataFrame, this will be
NA because we did not specify the data origin:
dataOrigin(sps)## [1] NA NA NAdataOrigin for sps_sciex, the
Spectra which was initialized with data from mzML files, in
contrast, returns the originating file names:
head(basename(dataOrigin(sps_sciex)))## [1] "7482fe40e87_7859" "7482fe40e87_7859" "7482fe40e87_7859" "7482fe40e87_7859"
## [5] "7482fe40e87_7859" "7482fe40e87_7859"The current data storage location of a spectrum can be retrieved with
the dataStorage variable, which will return an arbitrary
string for Spectra that use an in-memory backend or the
file where the data is stored for on-disk backends:
dataStorage(sps)## [1] "<memory>" "<memory>" "<memory>"
head(basename(dataStorage(sps_sciex)))## [1] "7482fe40e87_7859" "7482fe40e87_7859" "7482fe40e87_7859" "7482fe40e87_7859"
## [5] "7482fe40e87_7859" "7482fe40e87_7859"Certain backends (such as the MsBackendMemory and
MsBackendDataFrame) support also additional peaks
variables. At present, these must already be present when the backend
gets initialized. In future a dedicated function allowing to add peaks
variables will be available. Below we thus first extract the full data
(including peaks variables) from the sps spectra object and
add a column "peak_anno" with peak annotations for
each individual peak. Importantly, for peak variables, a value needs to
be assigned to each individual peak, even it it is NA (the
lengths() of the new peak variable must match
lengths() of mz or intensity,
i.e. the number of peaks per spectrum).
## Extract the full data from a spectrum
spd <- spectraData(sps, columns = union(spectraVariables(sps),
peaksVariables(sps)))
## Add a new column with a *annotation* for each peak
spd$peak_anno <- list(c("a", NA_character_, "b", "c", "d"),
c("a", "b", "c", "d", "e", "f", "g"),
c("a", "b", "c", "d", "e", "f", "g", "h", "i"))
## lengths have to match:
lengths(spd$peak_anno)## [1] 5 7 9
lengths(spd$mz)## [1] 5 7 9The parameter peaksVariables() (currently only available
for the backendInitialize() method of
MsBackendMemory and MsBackendDataFrame) allows
to define which of the columns from an input data contain peaks
variables (in our case "mz", "intensity" and
the additional "peak_anno" column).
sps2 <- Spectra(spd, backend = MsBackendMemory(),
peaksVariables = c("mz", "intensity", "peak_anno"))
peaksVariables(sps2)## [1] "mz" "intensity" "peak_anno"Full peak data can be extracted with the peaksData()
function that has a second parameter columns allowing to
define which peak variables to return. Below we extract the peak data
for the second spectrum.
peaksData(sps2, columns = peaksVariables(sps2))[[2L]]## mz intensity peak_anno
## 1 83.10 6.685 a
## 2 96.12 4.381 b
## 3 97.14 3.022 c
## 4 109.14 16.708 d
## 5 124.08 100.000 e
## 6 125.10 4.565 f
## 7 170.16 40.643 gWe can also use the peaksData() function to extract the
values for individual peak variables.
## Peak annotations for the first spectrum
peaksData(sps2, "peak_anno")[[1L]]## peak_anno
## 1 a
## 2 <NA>
## 3 b
## 4 c
## 5 d
## Peak annotations for the second spectrum
peaksData(sps2, "peak_anno")[[2L]]## peak_anno
## 1 a
## 2 b
## 3 c
## 4 d
## 5 e
## 6 f
## 7 gPeak variables can also be extracted using the $
method:
sps2$peak_anno## [[1]]
## [1] "a" NA "b" "c" "d"
##
## [[2]]
## [1] "a" "b" "c" "d" "e" "f" "g"
##
## [[3]]
## [1] "a" "b" "c" "d" "e" "f" "g" "h" "i"Similar to spectra variables it is also possible to replace values
for existing peaks variables using the
$<- function.
Filtering, aggregating and merging spectra data
Various functions are available to filter, subset and merge
Spectra objects. These can be generally subdivided into
functions that subset or filter spectra data and operations
that filter mass peak data. A third category of function allows
to aggregate data within a Spectra or to merge and combine
multiple Spectra objects into one. Functions of the various
categories are described in the following subsections. Please refer to
the function’s documentation for more details and information.
Filter spectra data
These functions comprise subset operations that reduce the total
number of spectra in a Spectra object as well as filter
functions that reduce the content of the Spectra’s spectra
data (i.e. the content of its spectraVariables()). These
functions thus don’t change or affect the mass peaks data of the
Spectra’s individual spectra.
-
[: operation to reduce aSpectraobject to selected elements. -
dropNaSpectraVariables(): dropsspectraVariables()that contain only missing values. The function returns aSpectraobject with the same number of elements, but with eventually fewer spectra variables. -
filterAcquisitionNum(): retains spectra with certain acquisition numbers. -
filterDataOrigin(): subsets to spectra from specific origins. -
filterDataStorage(): subsets to spectra from certain data storage files. -
filterEmptySpectra(): removes spectra without mass peaks. -
filterIsolationWindow(): keeps spectra with the providedmzin their isolation window (m/z range). -
filterMsLevel(): filters by MS level. -
filterPolarity(): filters by polarity. -
filterPrecursorCharge(): retains (MSn) spectra with specified precursor charge(s). -
filterPrecursorIsotopes(): identifies precursor ions (from fragment spectra) that could represent isotopes of the same molecule. For each of these spectra groups only the spectrum of the monoisotopic precursor ion is returned. MS1 spectra are returned without filtering. -
filterPrecursorMaxIntensity(): filters spectra keeping, for groups of spectra with similar precursor m/z, the one spectrum with the highest precursor intensity. All MS1 spectra are returned without filtering. -
filterPrecursorMzRange(): retains (MSn) spectra with a precursor m/z within the provided m/z range. -
filterPrecursorMzValues((): retains (MSn) spectra with precursor m/z value matching the provided value(s) considering also atoleranceandppm. -
filterPrecursorScan(): retains (parent and children) scans of an acquisition number. -
filterRanges(): filters aSpectraobject based on (multiple) user defined numeric ranges for one or more available (numeric) spectra variables. -
filterRt(): filters based on retention time range. -
filterValues(): filters aSpectraobject based on similarities of numeric values of one or more available spectra variables. -
selectSpectraVariables(): reduces the (spectra) data within the object to the selected spectra variables.
Filter or aggregate mass peak data
These function filter or aggregate the mass peak data
(peaksData()) of each spectrum in a Spectra
without changing the total number of spectra.
-
combinePeaks(): groups peaks within each spectrum based on similarity of their m/z values and combines these into a single peak per peak group. -
deisotopeSpectra(): deisotopes each individual spectrum keeping only the monoisotopic peak for peaks groups of potential isotopologues. -
filterFourierTransformArtefacts(): removes (Orbitrap) fast fourier transform artifact peaks from spectra. -
filterIntensity(): filter each spectrum keeping only peaks with intensities meeting certain criteria. -
filterMzRange(): filters mass peaks keeping (or removing) those with an m/z within the provided m/z range. -
filterMzValues(): filters mass peaks within each spectrum keeping (or removing) those with an m/z matching the provided value(s). -
filterPeaksRanges(): filters mass peaks using any set of range-based filters on numeric spectra or peaks variables. -
filterPrecursorPeaks(): removes peaks with either an m/z value matching the precursor m/z of the respective spectrum (with parametermz = "==") or peaks with an m/z value larger or equal to the precursor m/z (with parametermz = ">="). -
reduceSpectra(): filters individual spectra keeping only the largest peak for groups of peaks with similar m/z values.
Merging, aggregating and splitting
-
c(): combine severalSpectrainto a singleSpectraobject. -
combineSpectra(): allows to combine the MS data from sets of spectra into a single spectrum per set. Thus, instead of filtering the data, this function aggregates it. -
joinSpectraData(): merge aDataFrameto the existing spectra data. -
split(): splits theSpectraobject based on a provided grouping factor.
Examples and use cases for filter operations
In this example, we use the filterValues() function to
retain spectra with a base peak m/z close to 100 (+/- 30 ppm) and a
retention time around 230 (+/- 5 s).
sps_sub <- filterValues(sps_sciex, spectraVariables = c("basePeakMZ", "rtime"),
values = c(123.089, 230), tolerance = c(0,5),
ppm = c(30, 0), match = "all")
length(sps_sub)## [1] 72Then, we demonstrate the usage of the filterRanges()
function to filter spectra based on ranges of values for variables such
as base peak m/z, peak count, and retention time.
sps_ranges <- filterRanges(sps_sciex,
spectraVariables = c("basePeakMZ","peaksCount",
"rtime"),
ranges = c(123.09,124, 3500, 3520, 259, 260),
match = "all")
length(sps_ranges)## [1] 1Only one spectrum matches all the ranges. Another option for
filterValues() and filterRanges() is to use
the parameter match = "any", which retains spectra that
match any one of the conditions instead of having to match all of them.
Let’s run the code once again but change the match parameter this
time:
sps_ranges <- filterRanges(sps_sciex,
spectraVariables = c("basePeakMZ",
"peaksCount", "rtime"),
ranges = c(123.09, 124, 3500, 3520, 259, 260),
match = "any")
length(sps_ranges)## [1] 473We can see many more spectra passed the filtering step this time.
In the example below we use specific functions to select all spectra measured in the second mzML file and subsequently filter them to retain spectra measured between 175 and 189 seconds in the measurement run.
fls <- unique(dataOrigin(sps_sciex))
file_2 <- filterDataOrigin(sps_sciex, dataOrigin = fls[2])
length(file_2)## [1] 931## [1] 50In addition, Spectra support also subsetting with
[. Below we perform the filtering above with [
-based subsetting.
sps_sciex[sps_sciex$dataOrigin == fls[2] &
sps_sciex$rtime >= 175 &
sps_sciex$rtime <= 189]## MSn data (Spectra) with 50 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 1 175.212 628
## 2 1 175.491 629
## 3 1 175.770 630
## 4 1 176.049 631
## 5 1 176.328 632
## ... ... ... ...
## 46 1 187.768 673
## 47 1 188.047 674
## 48 1 188.326 675
## 49 1 188.605 676
## 50 1 188.884 677
## ... 34 more variables/columns.
##
## file(s):
## 74890c8574_7860The equivalent using filter function is shown below, with the added benefit that the filtering is recorded in the processing slot.
sps_sciex |>
filterDataOrigin(fls[2]) |>
filterRt(c(175, 189))## MSn data (Spectra) with 50 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 1 175.212 628
## 2 1 175.491 629
## 3 1 175.770 630
## 4 1 176.049 631
## 5 1 176.328 632
## ... ... ... ...
## 46 1 187.768 673
## 47 1 188.047 674
## 48 1 188.326 675
## 49 1 188.605 676
## 50 1 188.884 677
## ... 34 more variables/columns.
##
## file(s):
## 74890c8574_7860
## Processing:
## Filter: select data origin(s) /github/home/.cache/R/ExperimentHub/74890c8574_7860 [Fri Jun 12 14:35:44 2026]
## Filter: select retention time [175..189] on MS level(s) [Fri Jun 12 14:35:44 2026]Note that the use of the filter functions might be more efficient for some backends, depending on their implementation, (e.g. database-based backends could translate the filter function into a SQL condition to perform the subsetting already within the database).
Multiple Spectra objects can also be combined into a
single Spectra with the c() or the
concatenateSpectra() function. The resulting
Spectra object will contain an union of the spectra
variables of the individual objects. Below we combine the
Spectra object sps with an additional object
containing another MS2 spectrum for Caffeine.
caf_df <- DataFrame(msLevel = 2L, name = "Caffeine",
id = "HMDB0001847",
instrument = "Agilent 1200 RRLC; Agilent 6520 QTOF",
splash = "splash10-0002-0900000000-413259091ba7edc46b87",
centroided = TRUE)
caf_df$mz <- list(c(110.0710, 138.0655, 138.1057, 138.1742, 195.9864))
caf_df$intensity <- list(c(3.837, 32.341, 0.84, 0.534, 100))
caf <- Spectra(caf_df)Next we combine the two objects.
sps <- concatenateSpectra(sps, caf)
sps## MSn data (Spectra) with 4 spectra in a MsBackendMemory backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 NA NA
## 2 2 NA NA
## 3 2 NA NA
## 4 2 NA NA
## ... 20 more variables/columns.
## Processing:
## Merge 2 Spectra into one [Fri Jun 12 14:35:44 2026]The resulting object contains now the data for all 4 MS2 spectra and an union of all spectra variables from both objects.
spectraVariables(sps)## [1] "msLevel" "rtime"
## [3] "acquisitionNum" "scanIndex"
## [5] "dataStorage" "dataOrigin"
## [7] "centroided" "smoothed"
## [9] "polarity" "precScanNum"
## [11] "precursorMz" "precursorIntensity"
## [13] "precursorCharge" "collisionEnergy"
## [15] "isolationWindowLowerMz" "isolationWindowTargetMz"
## [17] "isolationWindowUpperMz" "id"
## [19] "name" "splash"
## [21] "instrument"The second object had an additional spectra variable
instrument that was not present in sps and all the
spectra in this object will thus get a value of NA for this
variable.
sps$instrument## [1] NA
## [2] NA
## [3] NA
## [4] "Agilent 1200 RRLC; Agilent 6520 QTOF"Sometimes not all spectra variables might be required (e.g. also
because many of them are empty). This might be specifically interesting
also for Spectra containing the data from very large
experiments, because it can significantly reduce the object’s size in
memory. In such cases the selectSpectraVariables() function
can be used to retain only specified spectra variables.
Data manipulations
Some analyses require manipulation of the mass peak data (i.e. the m/z and/or intensity values). The available functions are:
-
addProcessing(): apply any arbitrary function to modify thepeaksData()of each spectrum. -
bin(): aggregates peaks in each spectrum into discrete m/z bins. -
neutralLoss(): create neutral loss versions of spectra. -
pickPeaks(): centroiding of spectrum data. -
replaceIntensitiesBelow(): replaces intensities below a specified threshold with a value.
-
scalePeaks(): scales peak intensities. -
shiftPeaks(): shifts peaks by an offset along m/z dimension. -
smooth(): smooths spectra.
One example would be to remove all peaks from a spectrum that have an
intensity lower than a certain threshold. Below we perform such an
operation with the replaceIntensitiesBelow() function to
replace peak intensities below 10 in each spectrum in sps
with a value of 0.
sps_rep <- replaceIntensitiesBelow(sps, threshold = 10, value = 0)As a result intensities below 10 were set to 0 for all peaks.
intensity(sps_rep)## NumericList of length 4
## [[1]] 0 47.494 0 100 13.24
## [[2]] 0 0 0 16.708 100 0 40.643
## [[3]] 0 0 0 0 0 0 0 100 40.994
## [[4]] 0 32.341 0 0 100Zero-intensity peaks (and peaks with missing intensities) can then be
removed with the filterIntensity() function specifying a
lower required intensity level or optionally also an upper intensity
limit.
sps_rep <- filterIntensity(sps_rep, intensity = c(0.1, Inf))
intensity(sps_rep)## NumericList of length 4
## [[1]] 47.494 100 13.24
## [[2]] 16.708 100 40.643
## [[3]] 100 40.994
## [[4]] 32.341 100The filterIntensity() supports also a user-provided
function to be passed with parameter intensity which would
allow e.g. to remove peaks smaller than the median peak intensity of a
spectrum. See examples in the ?filterIntensity help page
for details.
Note that any data manipulations on Spectra objects are
not immediately applied to the peak data. They are added to a so called
processing queue which is applied each time peak data is
accessed (with the peaksData(), mz() or
intensity() functions). Thanks to this processing queue
data manipulation operations are also possible for read-only
backends (e.g. mzML-file based backends or database-based backends). The
information about the number of such processing steps can be seen below
(next to Lazy evaluation queue).
sps_rep## MSn data (Spectra) with 4 spectra in a MsBackendMemory backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 NA NA
## 2 2 NA NA
## 3 2 NA NA
## 4 2 NA NA
## ... 20 more variables/columns.
## Lazy evaluation queue: 2 processing step(s)
## Processing:
## Merge 2 Spectra into one [Fri Jun 12 14:35:44 2026]
## Signal <= 10 in MS level(s) 2 set to 0 [Fri Jun 12 14:35:44 2026]
## Remove peaks with intensities outside [0.1, Inf] in spectra of MS level(s) 2. [Fri Jun 12 14:35:44 2026]It is possible to add also custom functions to the processing queue
of a Spectra object. Such a function must take a peaks
matrix as its first argument, have ... in the function
definition and must return a peaks matrix (a peaks matrix is a numeric
two-column matrix with the first column containing the peaks’ m/z values
and the second the corresponding intensities). Below we define a
function that divides the intensities of each peak by a value which can
be passed with argument y.
## Define a function that takes a matrix as input, divides the second
## column by parameter y and returns it. Note that ... is required in
## the function's definition.
divide_intensities <- function(x, y, ...) {
x[, 2] <- x[, 2] / y
x
}
## Add the function to the procesing queue
sps_2 <- addProcessing(sps_rep, divide_intensities, y = 2)
sps_2## MSn data (Spectra) with 4 spectra in a MsBackendMemory backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 NA NA
## 2 2 NA NA
## 3 2 NA NA
## 4 2 NA NA
## ... 20 more variables/columns.
## Lazy evaluation queue: 3 processing step(s)
## Processing:
## Merge 2 Spectra into one [Fri Jun 12 14:35:44 2026]
## Signal <= 10 in MS level(s) 2 set to 0 [Fri Jun 12 14:35:44 2026]
## Remove peaks with intensities outside [0.1, Inf] in spectra of MS level(s) 2. [Fri Jun 12 14:35:44 2026]Object sps_2 has now 3 processing steps in its lazy
evaluation queue. Calling intensity() on this object will
now return intensities that are half of the intensities of the original
objects sps.
intensity(sps_2)## NumericList of length 4
## [[1]] 23.747 50 6.62
## [[2]] 8.354 50 20.3215
## [[3]] 50 20.497
## [[4]] 16.1705 50
intensity(sps_rep)## NumericList of length 4
## [[1]] 47.494 100 13.24
## [[2]] 16.708 100 40.643
## [[3]] 100 40.994
## [[4]] 32.341 100Alternatively we could define a function that returns the maximum
peak from each spectrum (note: we use the unname() function
to remove any names from the results):
max_peak <- function(x, ...) {
unname(x[which.max(x[, 2]), , drop = FALSE])
}
sps_2 <- addProcessing(sps_rep, max_peak)
lengths(sps_2)## [1] 1 1 1 1
intensity(sps_2)## NumericList of length 4
## [[1]] 100
## [[2]] 100
## [[3]] 100
## [[4]] 100Each spectrum in sps_2 thus contains only a single peak.
The parameter spectraVariables of the
addProcessing() function allows in addition to define
spectra variables that should be passed (in addition to the peaks
matrix) to the user-provided function. This would enable for example to
calculate neutral loss spectra from a Spectra by
subtracting the precursor m/z from each m/z of a spectrum (note that
there would also be a dedicated neutralLoss() function to
perform this operation more efficiently - or the
shiftPeaks() function could be used too).
Our toy example does not have precursor m/z values defined, thus we
first set them to arbitrary values. Then we define a function
neutral_loss that calculates the difference between the
precursor m/z and the fragment peak’s m/z. In addition we need to ensure
the peaks in the resulting spectra are ordered by (the delta) m/z
values. Note that, in order to be able to access the precursor m/z of
the spectrum within our function, we have to add a parameter to the
function that has the same name as the spectrum variable we want to
access (in our case precursorMz).
sps_rep$precursorMz <- c(170.5, 170.5, 195.1, 195.1)
neutral_loss <- function(x, precursorMz, ...) {
x[, "mz"] <- precursorMz - x[, "mz"]
x[order(x[, "mz"]), , drop = FALSE]
}We have then to call addProcessing() with
spectraVariables = "precursorMz" to specify that this
spectra variable is passed along to our function.
sps_3 <- addProcessing(sps_rep, neutral_loss,
spectraVariables = "precursorMz")
mz(sps_rep)## NumericList of length 4
## [[1]] 124.2 170.16 170.52
## [[2]] 109.14 124.08 170.16
## [[3]] 138.0662 195.0876
## [[4]] 138.0655 195.9864
mz(sps_3)## NumericList of length 4
## [[1]] -0.0200000000000102 0.340000000000003 46.3
## [[2]] 0.340000000000003 46.42 61.36
## [[3]] 0.0123999999999853 57.0338
## [[4]] -0.886400000000009 57.0345As we can see, the precursor m/z was subtracted from each m/z of the
respective spectrum. A better version of the function, that only
calculates neutral loss spectra for MS level 2 spectra would be the
neutral_loss function below. Since we are accessing also
the spectrum’s MS level we have to call addProcessing()
adding also the spectra variable msLevel to the
spectraVariables parameter. Note however that the
msLevel spectra variable is by default
renamed to spectrumMsLevel prior passing it to the
function. We have thus to use a parameter called
spectrumMsLevel in the neutral_loss function
instead of msLevel.
neutral_loss <- function(x, spectrumMsLevel, precursorMz, ...) {
if (spectrumMsLevel == 2L) {
x[, "mz"] <- precursorMz - x[, "mz"]
x <- x[order(x[, "mz"]), , drop = FALSE]
}
x
}
sps_3 <- addProcessing(sps_rep, neutral_loss,
spectraVariables = c("msLevel", "precursorMz"))
mz(sps_3)## NumericList of length 4
## [[1]] -0.0200000000000102 0.340000000000003 46.3
## [[2]] 0.340000000000003 46.42 61.36
## [[3]] 0.0123999999999853 57.0338
## [[4]] -0.886400000000009 57.0345Using the same concept it would also be possible to provide any
spectrum-specific user-defined value to the processing function. This
variable could simply be added first as a new spectra variable to the
Spectra object and then this variable could be passed along
to the function in the same way we passed the precursor m/z to our
function above.
Another example for spectra processing potentially helpful for
spectral matching against reference fragment spectra libraries would be
a function that removes fragment peaks with an m/z matching the
precursor m/z of a spectrum. Below we define such a function that takes
the peaks matrix and the precursor m/z as input and evaluates with the
closest() function from the MsCoreUtils
whether the spectrum contains peaks with an m/z value matching the one
of the precursor (given tolerance and ppm).
The returned peaks matrix contains all peaks except those matching the
precursor m/z.
##
## Attaching package: 'MsCoreUtils'## The following objects are masked from 'package:Spectra':
##
## bin, entropy, smooth## The following object is masked from 'package:stats':
##
## smooth
remove_precursor <- function(x, precursorMz, tolerance = 0.1, ppm = 0, ...) {
if (!is.na(precursorMz)) {
keep <- is.na(closest(x[, "mz"], precursorMz, tolerance = tolerance,
ppm = ppm, .check = FALSE))
x[keep, , drop = FALSE]
} else x
}We can now again add this processing step to our Spectra
object. As a result, peaks matching the precursor m/z (with
tolerance = 0.1 and ppm = 0) will be
removed.
sps_4 <- addProcessing(sps_rep, remove_precursor,
spectraVariables = "precursorMz")
peaksData(sps_4) |> as.list()## [[1]]
## mz intensity
## [1,] 124.20 47.494
## [2,] 170.16 100.000
##
## [[2]]
## mz intensity
## [1,] 109.14 16.708
## [2,] 124.08 100.000
## [3,] 170.16 40.643
##
## [[3]]
## mz intensity
## [1,] 138.0662 100
##
## [[4]]
## mz intensity
## [1,] 138.0655 32.341
## [2,] 195.9864 100.000As a reference, the original peak matrices are shown below.
## [[1]]
## mz intensity
## [1,] 124.20 47.494
## [2,] 170.16 100.000
## [3,] 170.52 13.240
##
## [[2]]
## mz intensity
## [1,] 109.14 16.708
## [2,] 124.08 100.000
## [3,] 170.16 40.643
##
## [[3]]
## mz intensity
## [1,] 138.0662 100.000
## [2,] 195.0876 40.994
##
## [[4]]
## mz intensity
## [1,] 138.0655 32.341
## [2,] 195.9864 100.000Note that we can also perform a more relaxed matching of m/z values
by passing a different value for tolerance to the
function:
sps_4 <- addProcessing(sps_rep, remove_precursor, tolerance = 0.6,
spectraVariables = "precursorMz")
peaksData(sps_4) |> as.list()## [[1]]
## mz intensity
## [1,] 124.2 47.494
##
## [[2]]
## mz intensity
## [1,] 109.14 16.708
## [2,] 124.08 100.000
##
## [[3]]
## mz intensity
## [1,] 138.0662 100
##
## [[4]]
## mz intensity
## [1,] 138.0655 32.341
## [2,] 195.9864 100.000Since all data manipulations above did not change the original
intensity or m/z values, it is possible to restore the original
data. This can be done with the reset() function which will
empty the lazy evaluation queue and call the reset() method
on the storage backend. Below we call reset() on the
sps_2 object and hence restore the data to its original
state.
## NumericList of length 4
## [[1]] 3.407 47.494 3.094 100 13.24
## [[2]] 6.685 4.381 3.022 16.708 100 4.565 40.643
## [[3]] 0.459 2.585 2.446 0.508 8.968 0.524 0.974 100 40.994
## [[4]] 3.837 32.341 0.84 0.534 100
intensity(sps)## NumericList of length 4
## [[1]] 3.407 47.494 3.094 100 13.24
## [[2]] 6.685 4.381 3.022 16.708 100 4.565 40.643
## [[3]] 0.459 2.585 2.446 0.508 8.968 0.524 0.974 100 40.994
## [[4]] 3.837 32.341 0.84 0.534 100Finally, for Spectra that use a writeable
backend, such as the MsBackendMemory,
MsBackendDataFrame or MsBackendHdf5Peaks, it
is possible to apply the processing queue to the peak data and write
that back to the data storage with the applyProcessing()
function. Below we use this to make all data manipulations on peak data
of the sps_rep object persistent.
length(sps_rep@processingQueue)## [1] 2
sps_rep <- applyProcessing(sps_rep)
length(sps_rep@processingQueue)## [1] 0
sps_rep## MSn data (Spectra) with 4 spectra in a MsBackendMemory backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 NA NA
## 2 2 NA NA
## 3 2 NA NA
## 4 2 NA NA
## ... 20 more variables/columns.
## Processing:
## Merge 2 Spectra into one [Fri Jun 12 14:35:44 2026]
## Signal <= 10 in MS level(s) 2 set to 0 [Fri Jun 12 14:35:44 2026]
## Remove peaks with intensities outside [0.1, Inf] in spectra of MS level(s) 2. [Fri Jun 12 14:35:44 2026]
## ...1 more processings. Use 'processingLog' to list all.Before applyProcessing() the lazy evaluation queue
contained 2 processing steps, which were then applied to the peak data
and written to the data storage. Note that calling
reset() after
applyProcessing() can no longer restore the
data.
Visualizing Spectra
The Spectra package provides the following functions to
visualize spectra data: - plotSpectra(): plot each spectrum
in Spectra in its own panel. -
plotSpectraOverlay(): plot multiple spectra into the
same plot.
Below we use plotSpectra() to plot the 4 spectra from
the sps object using their names (as provided in spectra
variable "name") as plot titles.
plotSpectra(sps, main = sps$name)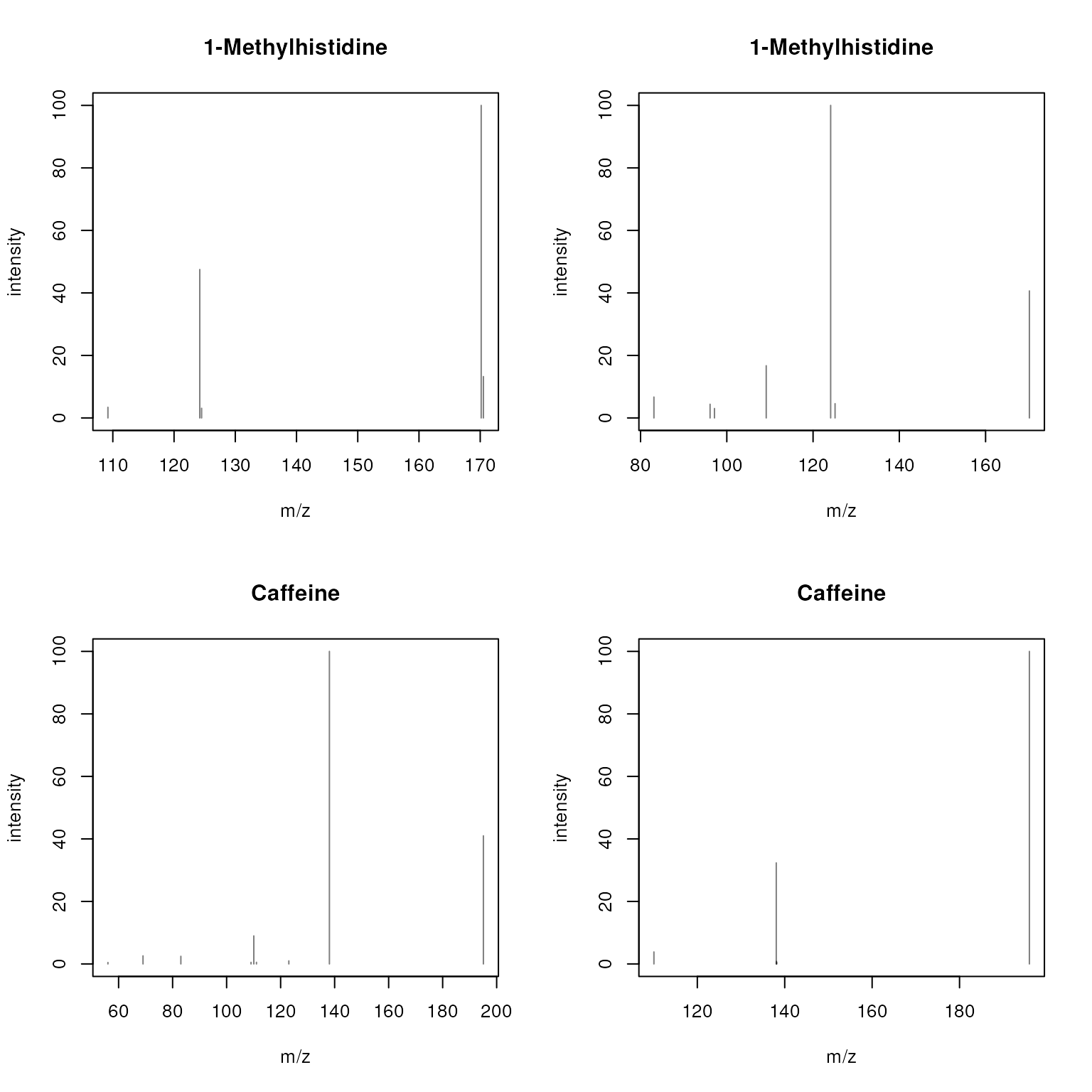
It is also possible to label individual peaks in each plot. Below we
use the m/z value of each peak as its label. In the example we define a
function that accesses information from each spectrum (z)
and returns a character for each peak with the text that
should be used as label. Parameters labelSrt,
labelPos and labelOffset define the rotation
of the label text and its position relative to the x and y coordinates
of the peak.
plotSpectra(sps, main = sps$name,
labels = function(z) lapply(mz(z), format, digits = 4),
labelSrt = -30, labelPos = 2, labelOffset = 0.1)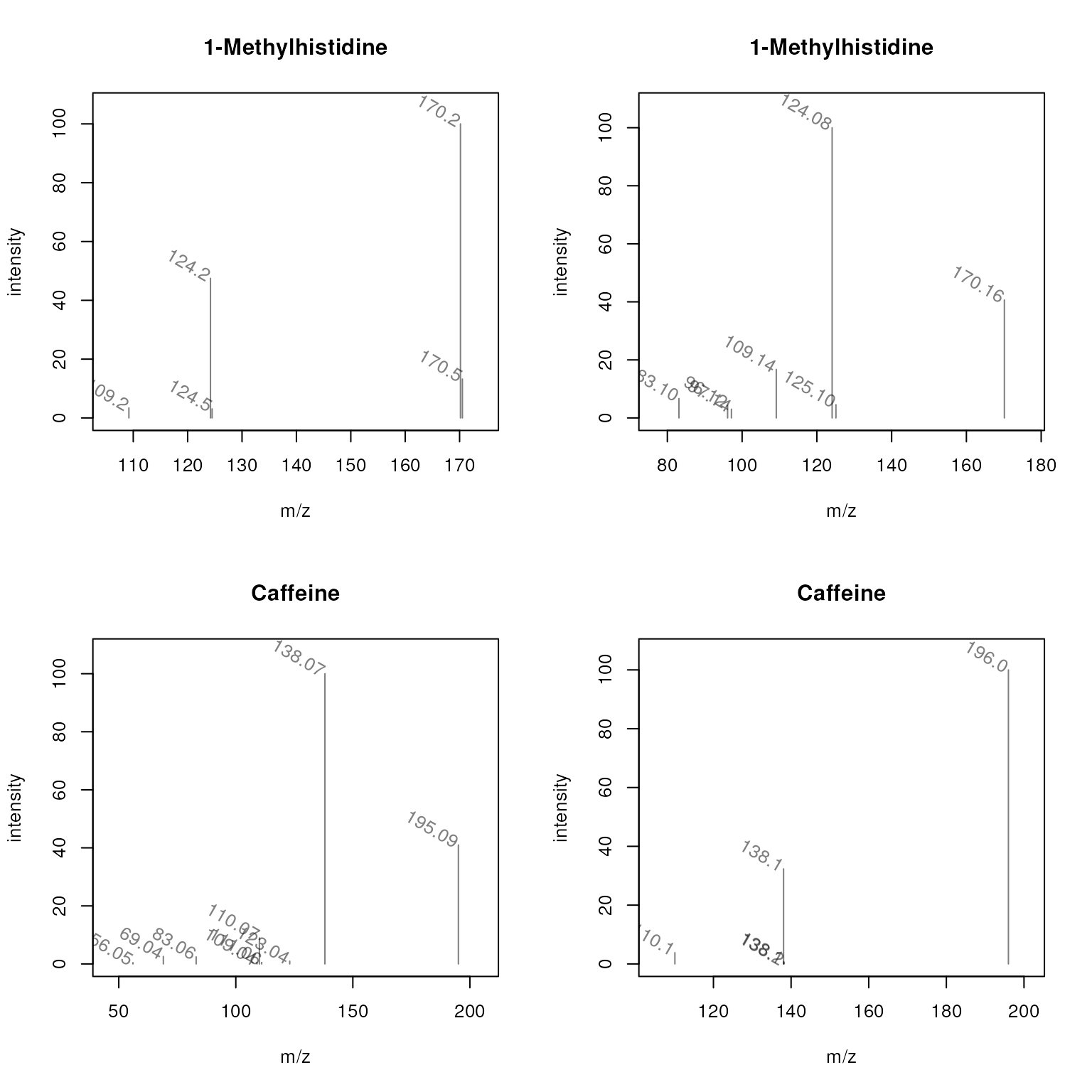
These plots are rather busy and for some peaks the m/z values are overplotted. Below we define a label function that will only indicate the m/z of peaks with an intensity higher than 30.
mzLabel = function(z) {
lapply(seq_along(mz(z)), function(i) {
lbls <- format(mz(z)[[i]], digits = 4)
lbls[intensity(z)[[i]] <= 30] <- ""
lbls
})
}
plotSpectra(sps, main = sps$name, labels = mzLabel,
labelSrt = -30, labelPos = 2, labelOffset = 0.1)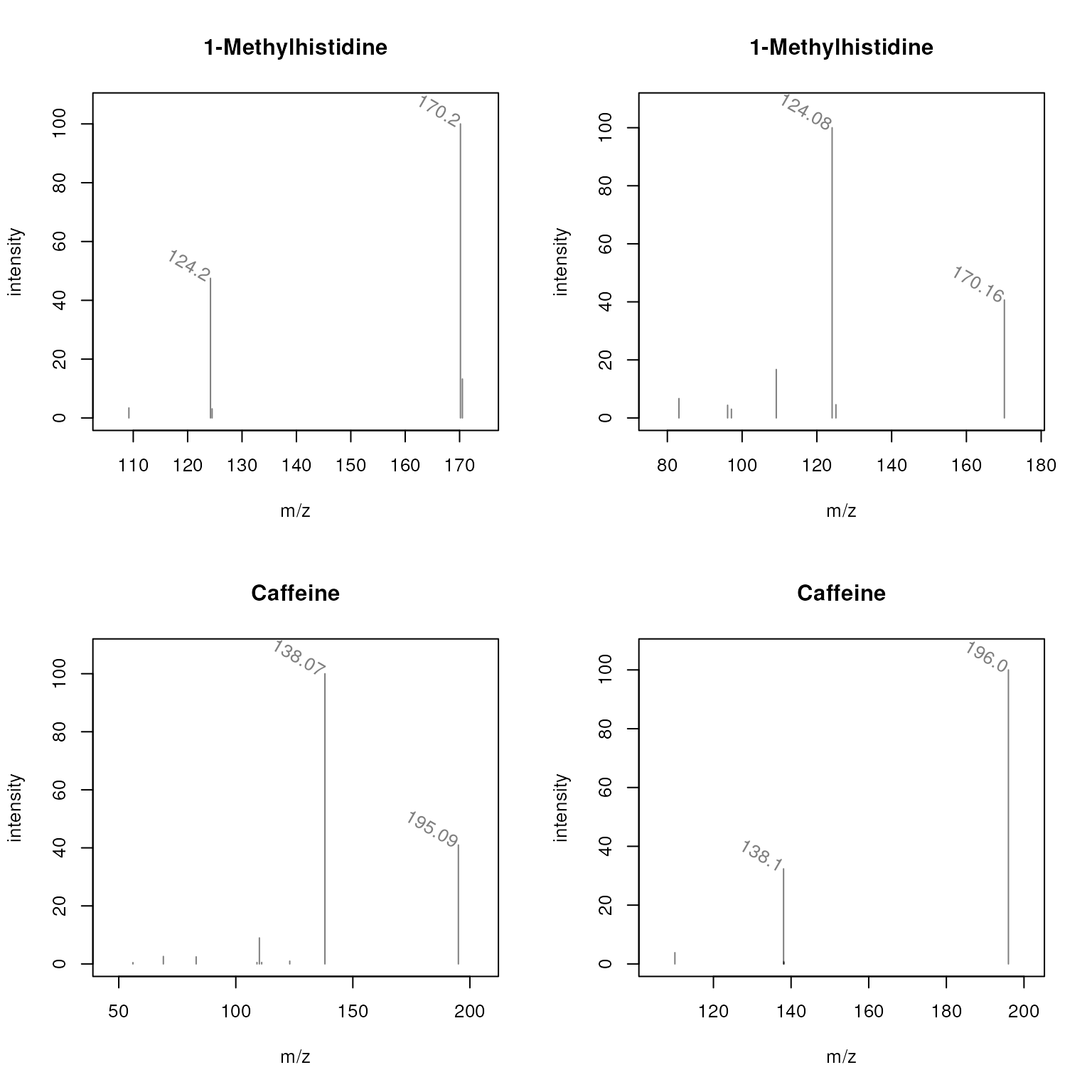
Sometimes it might be of interest to plot multiple spectra into the
same plot (e.g. to directly compare peaks from multiple
spectra). This can be done with plotSpectraOverlay() which
we use below to create an overlay-plot of our 4 example
spectra, using a different color for each spectrum.
cols <- c("#E41A1C80", "#377EB880", "#4DAF4A80", "#984EA380")
plotSpectraOverlay(sps, lwd = 2, col = cols)
legend("topleft", col = cols, legend = sps$name, pch = 15)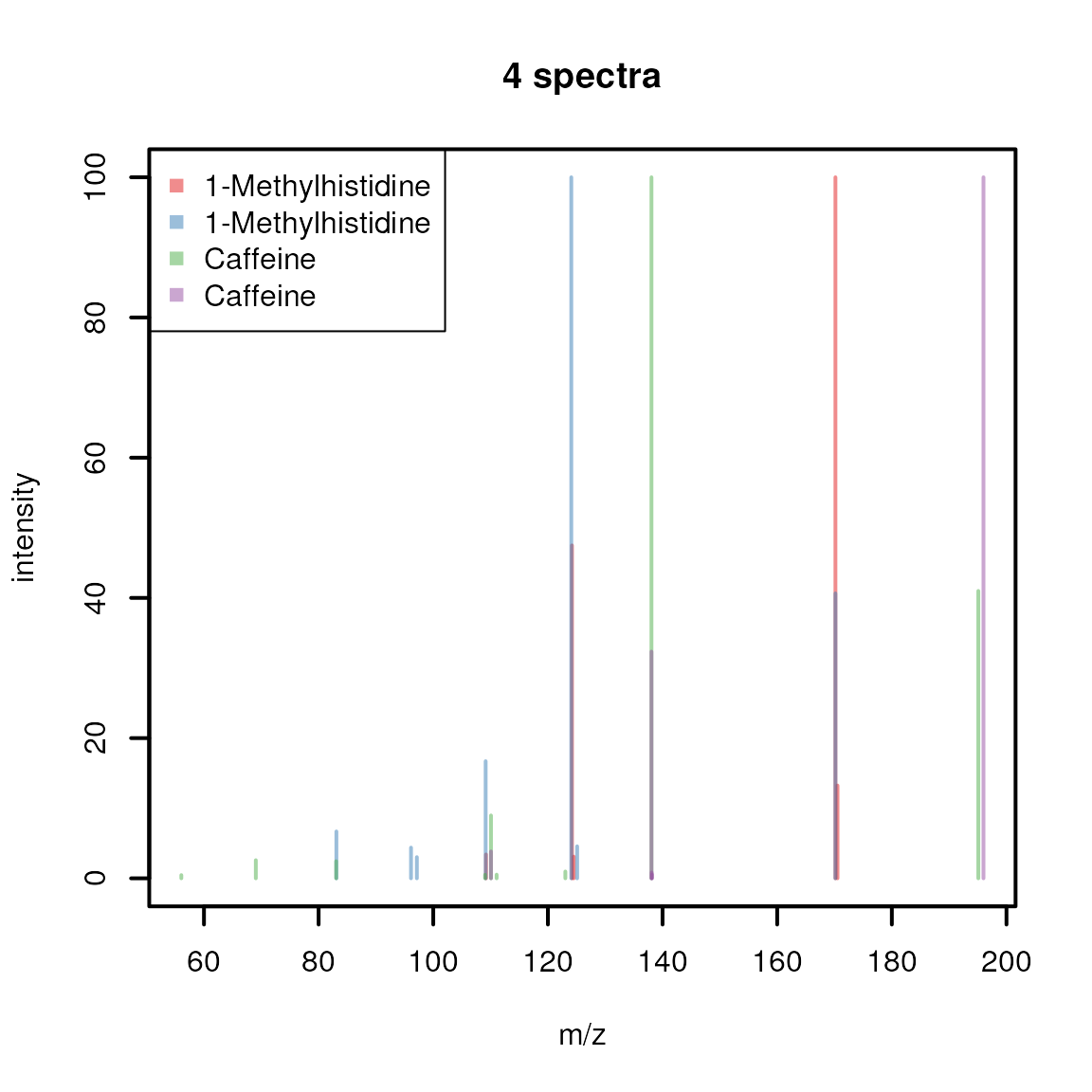
Lastly, plotSpectraMirror() allows to plot two spectra
against each other as a mirror plot which is ideal to visualize
spectra comparison results. Below we plot a spectrum of
1-Methylhistidine against one of Caffeine.
plotSpectraMirror(sps[1], sps[3])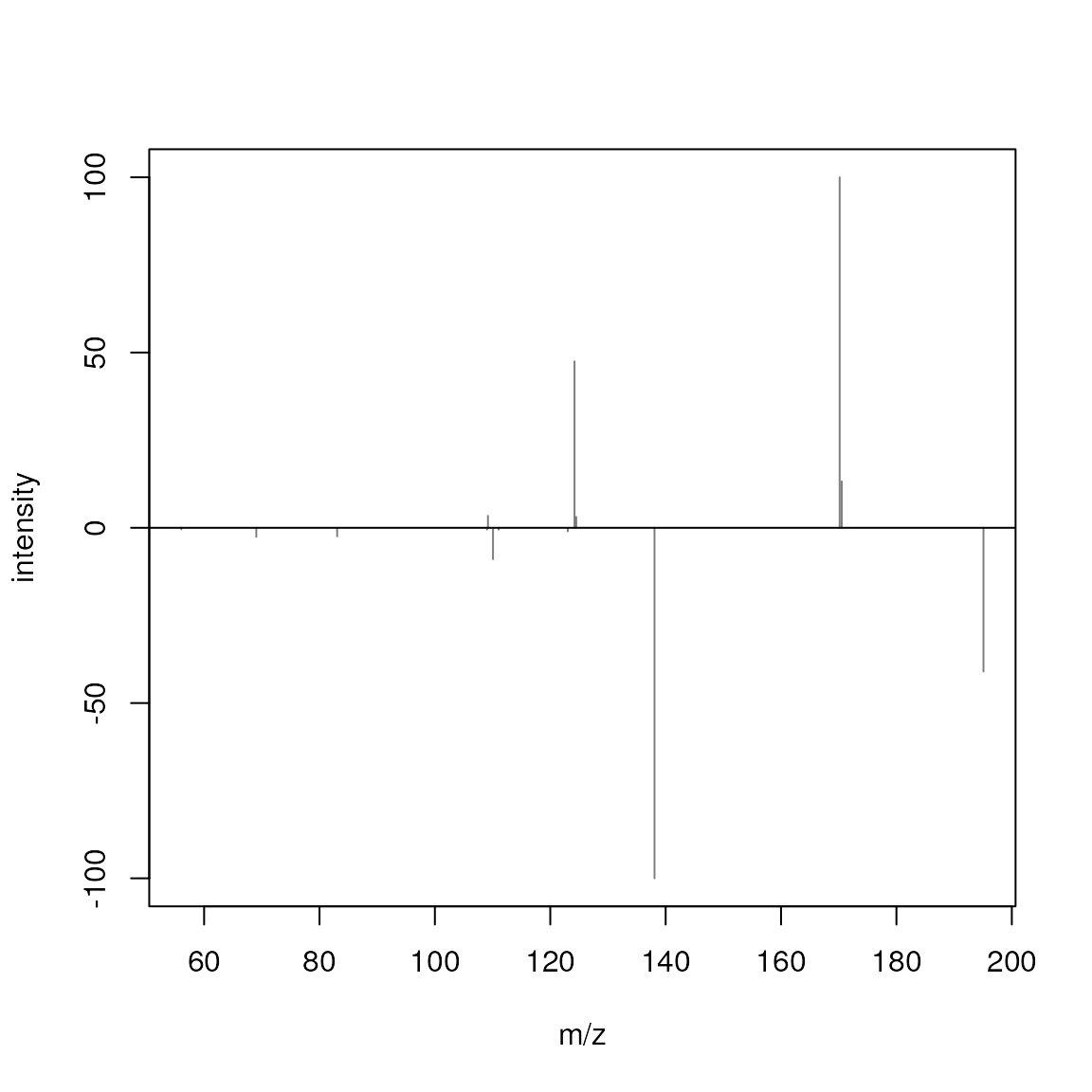
The upper panel shows the spectrum from 1-Methylhistidine, the lower
the one of Caffeine. None of the peaks of the two spectra match. Below
we plot the two spectra of 1-Methylhistidine and the two of Caffeine
against each other matching peaks with a ppm of 50.
par(mfrow = c(1, 2))
plotSpectraMirror(sps[1], sps[2], main = "1-Methylhistidine", ppm = 50)
plotSpectraMirror(sps[3], sps[4], main = "Caffeine", ppm = 50)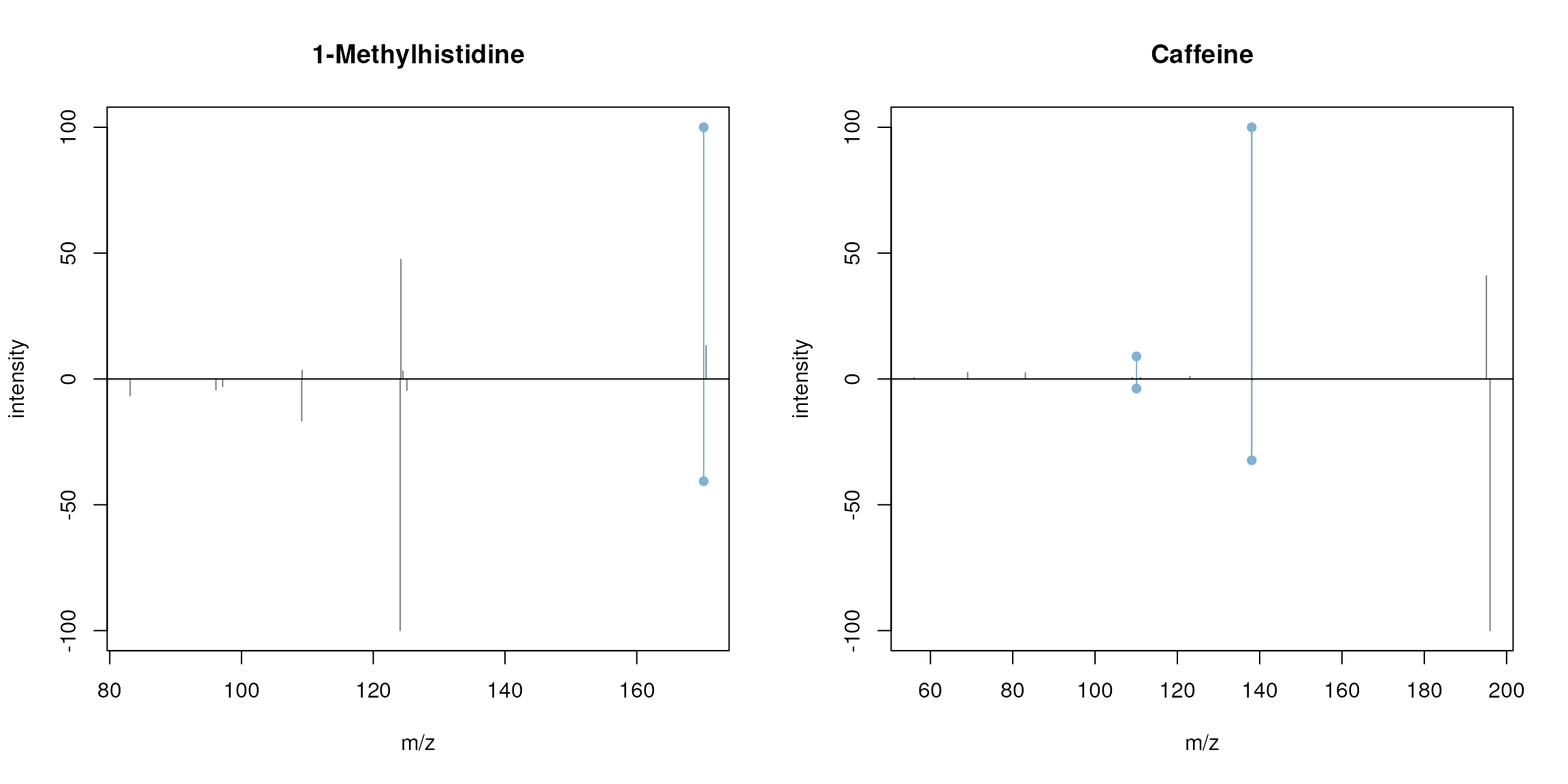
See also ?plotSpectra for more plotting options and
examples.
Aggregating spectra data
The Spectra package provides the
combineSpectra() function that allows to aggregate
multiple spectra into a single one. The main parameters of this function
are f, which defines the sets of spectra that should be
combined, and FUN, which allows to define the function that
performs the actual aggregation. The default aggregation function is
combinePeaksData() (see ?combinePeaksData for
details) that combines multiple spectra into a single spectrum with all
peaks from all input spectra (with additional paramter
peaks = "union"), or peaks that are present in a certain
proportion of input spectra (with parameter
peaks = "intersect"; parameter minProp allows
to define the minimum required proportion of spectra in which a peak
needs to be present. It is important to mention that, by default, the
function combines all mass peaks from all spectra with a similar m/z
value into a single, representative mass peak aggregating all their
intensities into one. To avoid the resulting intensity to be affected by
potential noise peaks it might be advised to first clean the
individual mass spectra using e.g. the combinePeaks() or
reduceSpectra() functions that first aggregate mass peaks
within each individual spectrum.
In this example we below we use combineSpectra() to
combine the spectra for 1-methylhistidine and caffeine into a single
spectrum for each compound. We use the spectra variable
$name, that contains the names of the compounds, to define
which spectra should be grouped together.
sps_agg <- combineSpectra(sps, f = sps$name)As a result, the 4 spectra got aggregated into two.
plotSpectra(sps_agg, main = sps_agg$name)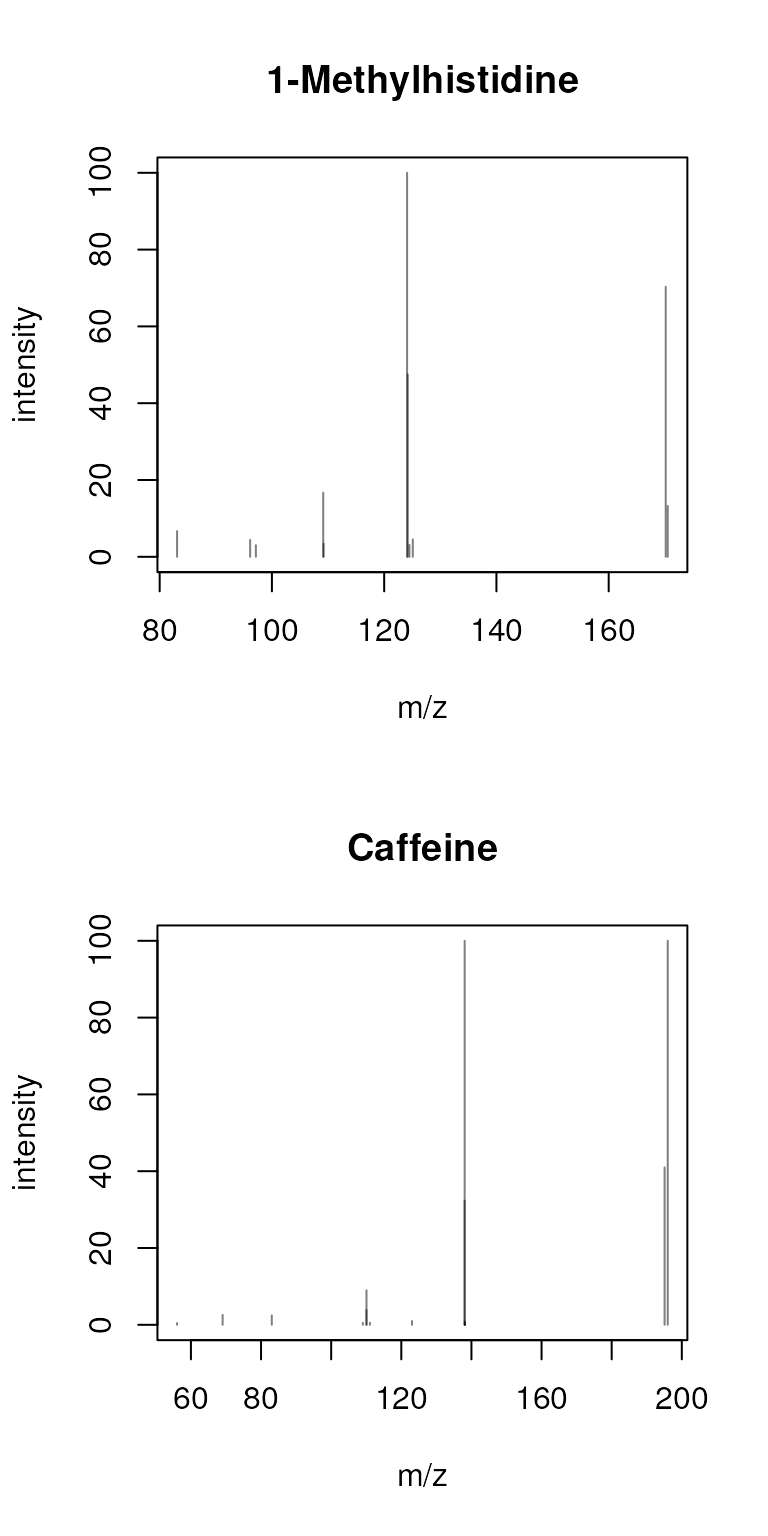
By default, all peaks present in all spectra are reported. As an
alternative, by specifying peaks = "intersect" and
minProp = 1, we could combine the spectra keeping only
peaks that are present in both input spectra.
sps_agg <- combineSpectra(sps, f = sps$name, peaks = "intersect", minProp = 1)
plotSpectra(sps_agg, main = sps_agg$name)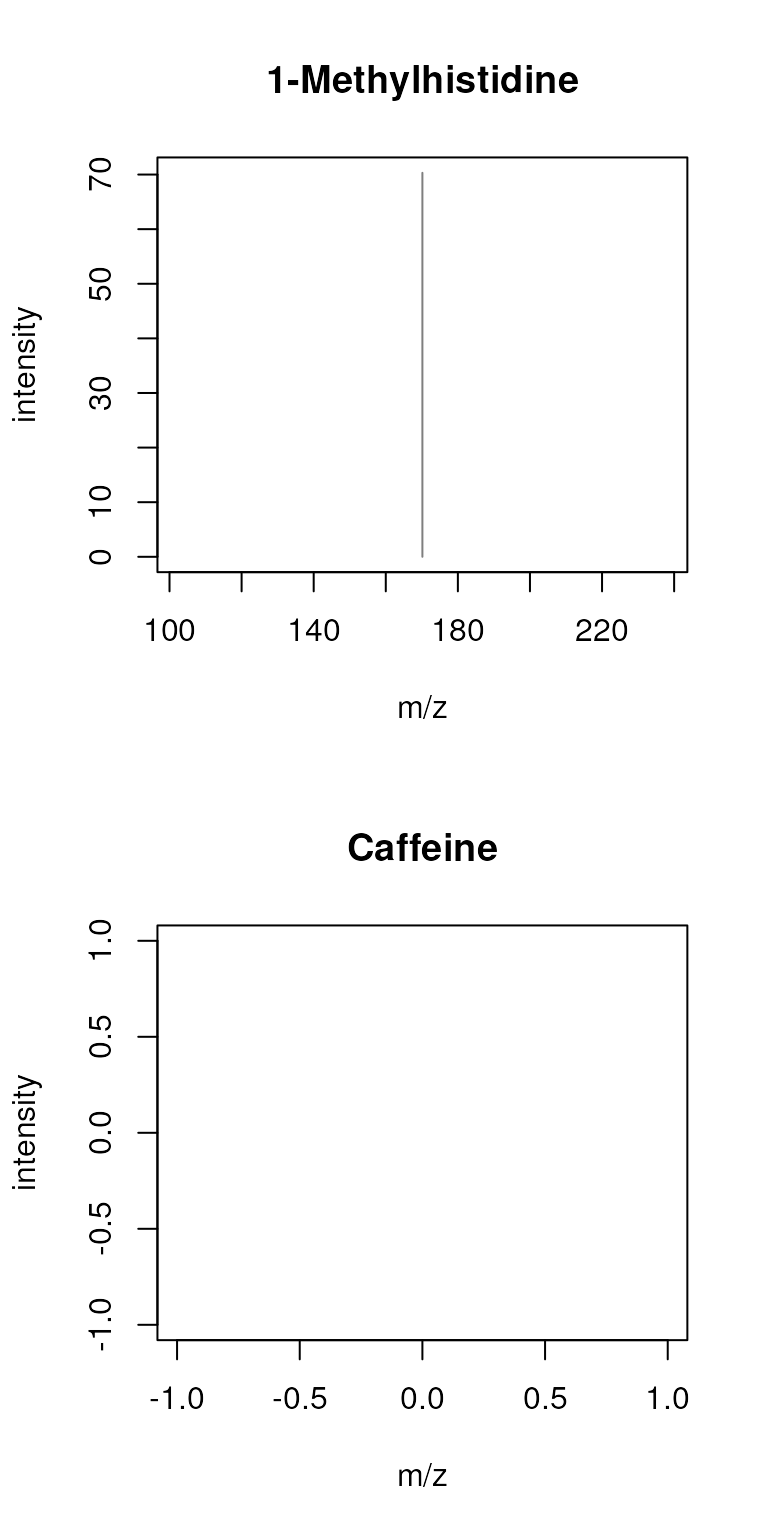
This results thus in a single peak for 1-methylhistidine and none for
caffeine - why? The reason for that is that the difference of the peaks’
m/z values is larger than the default tolerance used for the peak
grouping (the defaults for combinePeaksData() is
tolerance = 0 and ppm = 0). We could however
already see in the previous section that the reported peaks’ m/z values
have a larger measurement error (most likely because the fragment
spectra were measured on different instruments with different
precision). Thus, we next increase the tolerance and
ppm parameters to group also peaks with a larger difference
in their m/z values.
sps_agg <- combineSpectra(sps, f = sps$name, peaks = "intersect",
minProp = 1, tolerance = 0.2)
plotSpectra(sps_agg, main = sps_agg$name)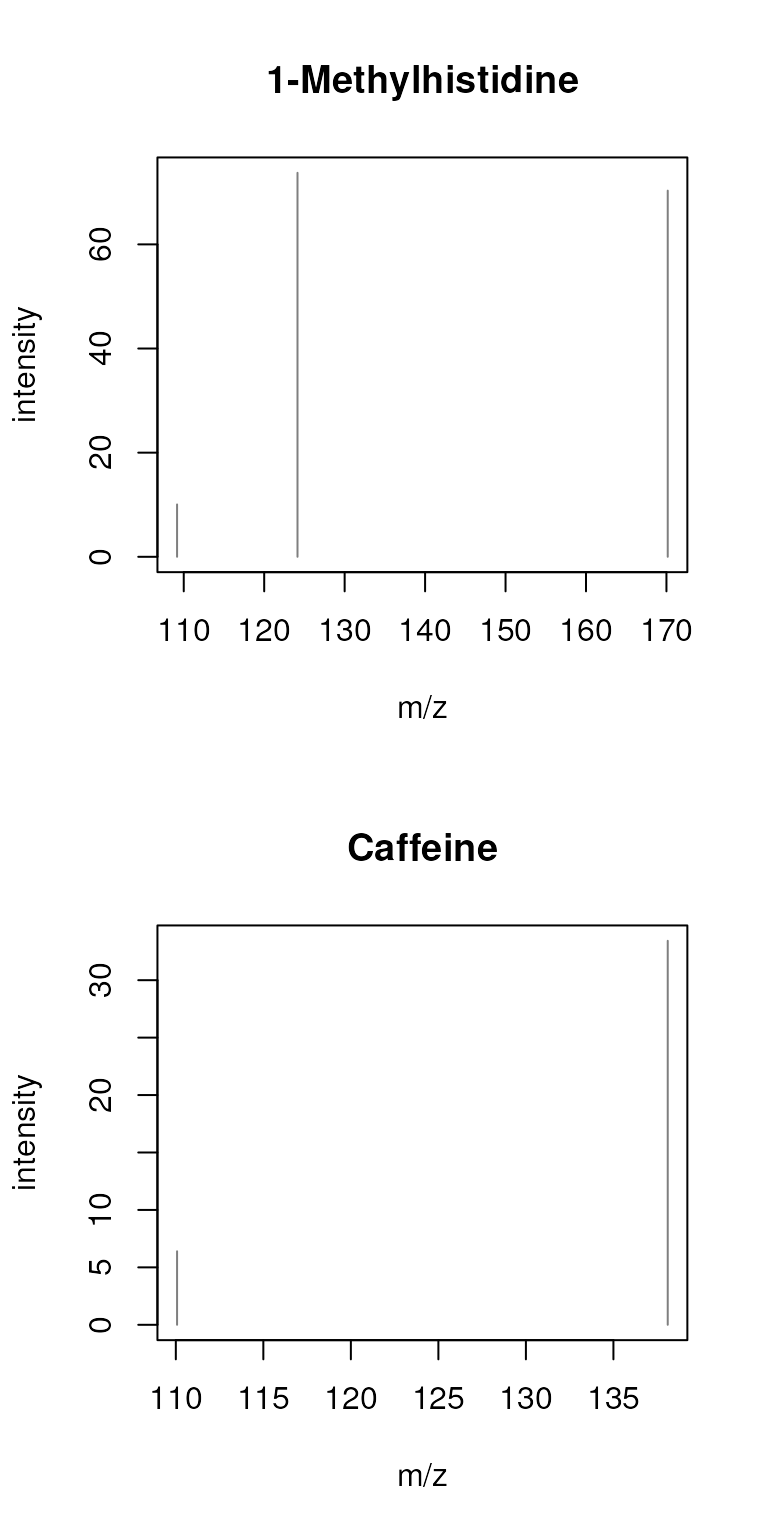
Whether in a real analysis we would be OK with such a large tolerance
is however questionable. Note: which m/z and intensity is reported for
the aggregated spectra can be defined with the parameters
intensityFun and mzFun of
combinePeaksData() (see ?combinePeaksData for
more information).
While the combinePeaksData() function is indeed helpful
to combine peaks from different spectra, the
combineSpectra() function would in addition also allow us
to provide our own, custom, peak aggregation function. As a simple
example, instead of combining the spectra, we would like to select one
of the input spectra as representative spectrum for grouped
input spectra. combineSpectra() supports any function that
takes a list of peak matrices as input and returns a single peak matrix
as output. We thus define below a function that calculates the total
signal (TIC) for each input peak matrix, and returns the one peak matrix
with the largest TIC.
#' function to select and return the peak matrix with the largest tic from
#' the provided list of peak matrices.
maxTic <- function(x, ...) {
tic <- vapply(x, function(z) sum(z[, "intensity"], na.rm = TRUE),
numeric(1))
x[[which.max(tic)]]
}We can now use this function with combineSpectra() to
select for each compound the spectrum with the largest TIC.
sps_agg <- combineSpectra(sps, f = sps$name, FUN = maxTic)
plotSpectra(sps_agg, main = sps_agg$name)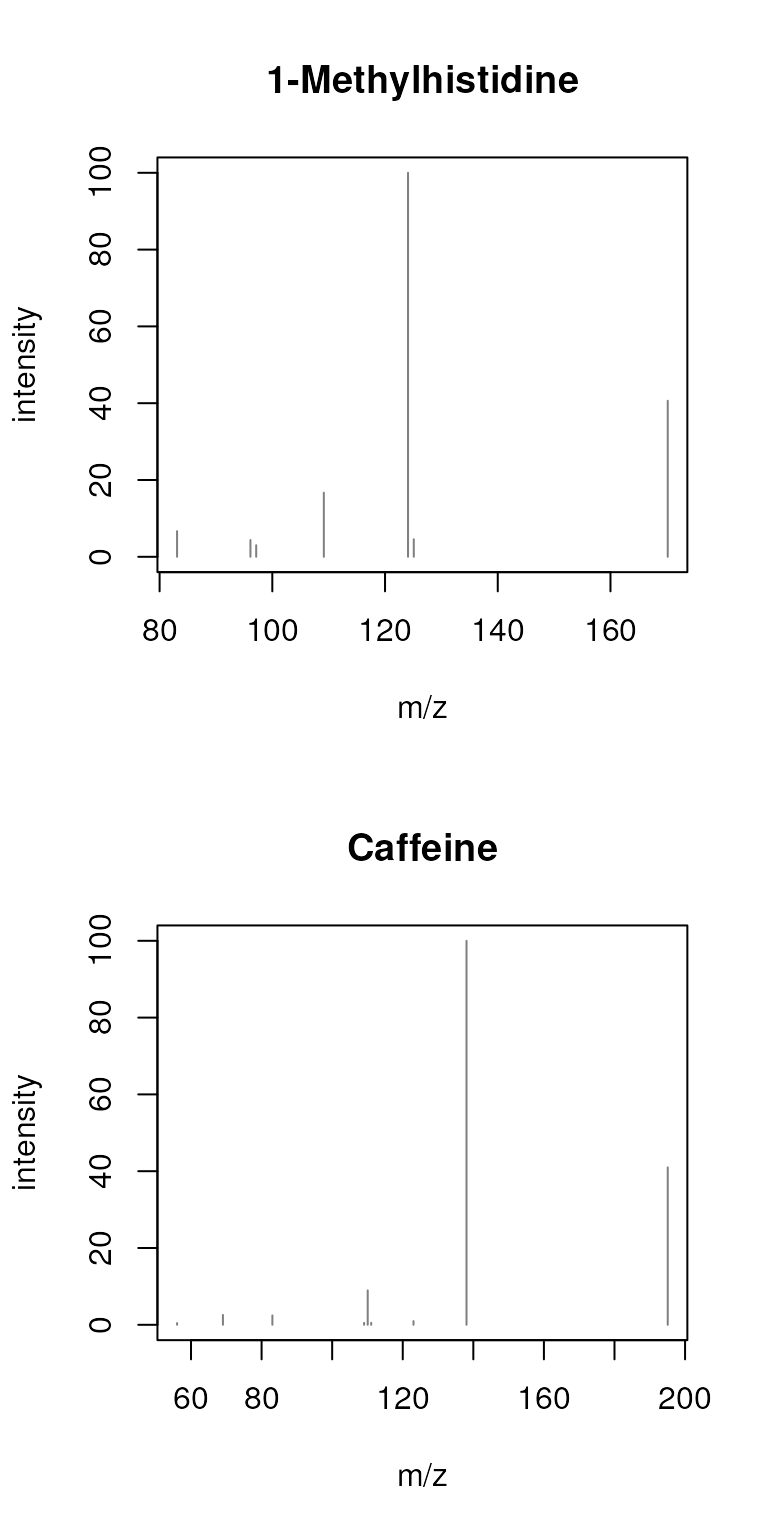
Comparing spectra
Spectra can be compared with the compareSpectra()
function, that allows to calculate similarities between spectra using a
variety of methods. compareSpectra() implements similarity
scoring as a two-step approach: first the peaks from the pair of spectra
that should be compared are matched (mapped) against each other and then
a similarity score is calculated on these. The MAPFUN
parameter of compareSpectra() defines the function to match
(or map) the peaks between the spectra and parameter FUN
specifies the function to calculate the similarity. By default,
compareSpectra() uses MAPFUN = joinPeaks (see
?joinPeaks for a description and alternative options) and
FUN = ndotproduct (the normalized dot-product spectra
similarity score). Parameters to configure these functions can be passed
to compareSpectra() as additional parameter (such as
e.g. ppm to define the m/z-relative tolerance for peak
matching in joinPeaks()).
Below we calculate pairwise similarities between all spectra in
sps accepting a 50 ppm difference of peaks’ m/z values for
being considered matching.
compareSpectra(sps, ppm = 50)## 1 2 3 4
## 1 1.0000000 0.1380817 0.0000000 0.0000000
## 2 0.1380817 1.0000000 0.0000000 0.0000000
## 3 0.0000000 0.0000000 1.0000000 0.1817149
## 4 0.0000000 0.0000000 0.1817149 1.0000000The resulting matrix provides the similarity scores from the pairwise
comparison. As expected, the first two and the last two spectra are
similar, albeit only moderately, while the spectra from
1-Methylhistidine don’t share any similarity with those of Caffeine.
Similarities between Spectra objects can be
calculated with calls in the form of compareSpectra(a, b)
with a and b being the two
Spectra objects to compare. As a result a n x m
matrix will be returned with n (rows) being the spectra in
a and m (columns) being the spectra in
b.
By setting parameter matchedPeaksCount = TRUE in
addition to the similarity scores, also the number of matching peaks
between the compared spectra are returned. The result is then a
3-dimensional array with the similarity scores in the first
matrix in z dimension ([, , 1]) and the number
of matching peaks in the second matrix
([, , 2]):
sim <- compareSpectra(sps, sps, ppm = 40, matchedPeaksCount = TRUE)
#' The similarity scores:
sim[, , 1L]## 1 2 3 4
## 1 1.0000000 0.1380817 0.0000000 0.0000000
## 2 0.1380817 1.0000000 0.0000000 0.0000000
## 3 0.0000000 0.0000000 1.0000000 0.1817149
## 4 0.0000000 0.0000000 0.1817149 1.0000000
#' The number of matching peaks:
sim[, , 2L]## 1 2 3 4
## 1 5 1 0 0
## 2 1 7 0 0
## 3 0 0 9 2
## 4 0 0 2 5The above similarity was calculated with the default (normalized)
dot-product, but also other similarity scores can be used instead.
Either one of the other metrics provided by the MsCoreUtils
could be used (see ?MsCoreUtils::distance for a list of
available options) or any other external or user-provided similarity
scoring function. As an example, we use below the spectral entropy
similarity score introduced in (Y et al.
2021) and provided with the msentropy
package. Since this msentropy_similarity() function
performs also the mapping of the peaks between the compared spectra
internally (along with some spectra cleaning), we have to disable that
in the compareSpectra() function using
MAPFUN = joinPeaksNone. To configure the similarity scoring
we can pass all additional parameters of the
msentropy_similarity() (see
?msentropy_similarity) to the compareSpectra()
call. We use ms2_tolerance_in_ppm = 50 to set the tolerance
for m/z-relative peak matching (equivalent to ppm = 50 used
above) and ms2_tolerance_in_da = -1 to disable absolute
tolerance matching.
## Loading required package: Rcpp
compareSpectra(sps, MAPFUN = joinPeaksNone, FUN = msentropy_similarity,
ms2_tolerance_in_ppm = 50, ms2_tolerance_in_da = -1)## 1 2 3 4
## 1 1.0000000 0.3002225 0.0000000 0.0000000
## 2 0.3002225 1.0000000 0.0000000 0.0000000
## 3 0.0000000 0.0000000 1.0000000 0.5144764
## 4 0.0000000 0.0000000 0.5144764 1.0000000Note also that GNPS-like scores can be calculated with
MAPFUN = joinPeaksGnps and
FUN = MsCoreUtils::gnps. For additional information and
examples see also (Rainer et al. 2022) or
the SpectraTutorials
tutorial.
Another way of comparing spectra would be to bin the spectra
and to cluster them based on similar intensity values. Spectra binning
ensures that the binned m/z values are comparable across all spectra.
Below we bin our spectra using a bin size of 0.1 (i.e. all peaks with an
m/z smaller than 0.1 are aggregated into one binned peak. Below, we
explicitly set zero.rm = FALSE to retain all bins generated
by the function, including those with an intensity of zero.
sps_bin <- Spectra::bin(sps, binSize = 0.1, zero.rm = FALSE)All spectra will now have the same number of m/z values.
lengths(sps_bin)## [1] 1400 1400 1400 1400Most of the intensity values for these will however be 0 (because in the original spectra no peak for the respective m/z bin was present).
intensity(sps_bin)## NumericList of length 4
## [[1]] 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
## [[2]] 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
## [[3]] 0.459 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 40.994 0 0 0 0 0 0 0 0 0
## [[4]] 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 100We’re next creating an intensity matrix for our Spectra
object, each row being one spectrum and columns representing the binned
m/z values.
We can now identify those columns (m/z bins) with only 0s across all spectra and remove these.
zeros <- colSums(intmat) == 0
intmat <- intmat[, !zeros]
intmat## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12]
## [1,] 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 3.407 0.000 0.000 0.000
## [2,] 0.000 0.000 0.000 6.685 4.381 3.022 0.000 16.708 0.000 0.000 0.000 0.000
## [3,] 0.459 2.585 2.446 0.000 0.000 0.000 0.508 0.000 0.000 8.968 0.524 0.974
## [4,] 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 3.837 0.000 0.000
## [,13] [,14] [,15] [,16] [,17] [,18] [,19] [,20] [,21] [,22]
## [1,] 0 47.494 3.094 0.000 0.000 0.000 100.000 13.24 0.000 0
## [2,] 100 0.000 0.000 4.565 0.000 0.000 40.643 0.00 0.000 0
## [3,] 0 0.000 0.000 0.000 100.000 0.000 0.000 0.00 40.994 0
## [4,] 0 0.000 0.000 0.000 32.341 1.374 0.000 0.00 0.000 100The associated m/z values for the bins can be extracted with
mz() from the binned Spectra object. Below we
use these as column names for the intensity matrix.
This intensity matrix could now for example be used to cluster the spectra based on their peak intensities.
heatmap(intmat)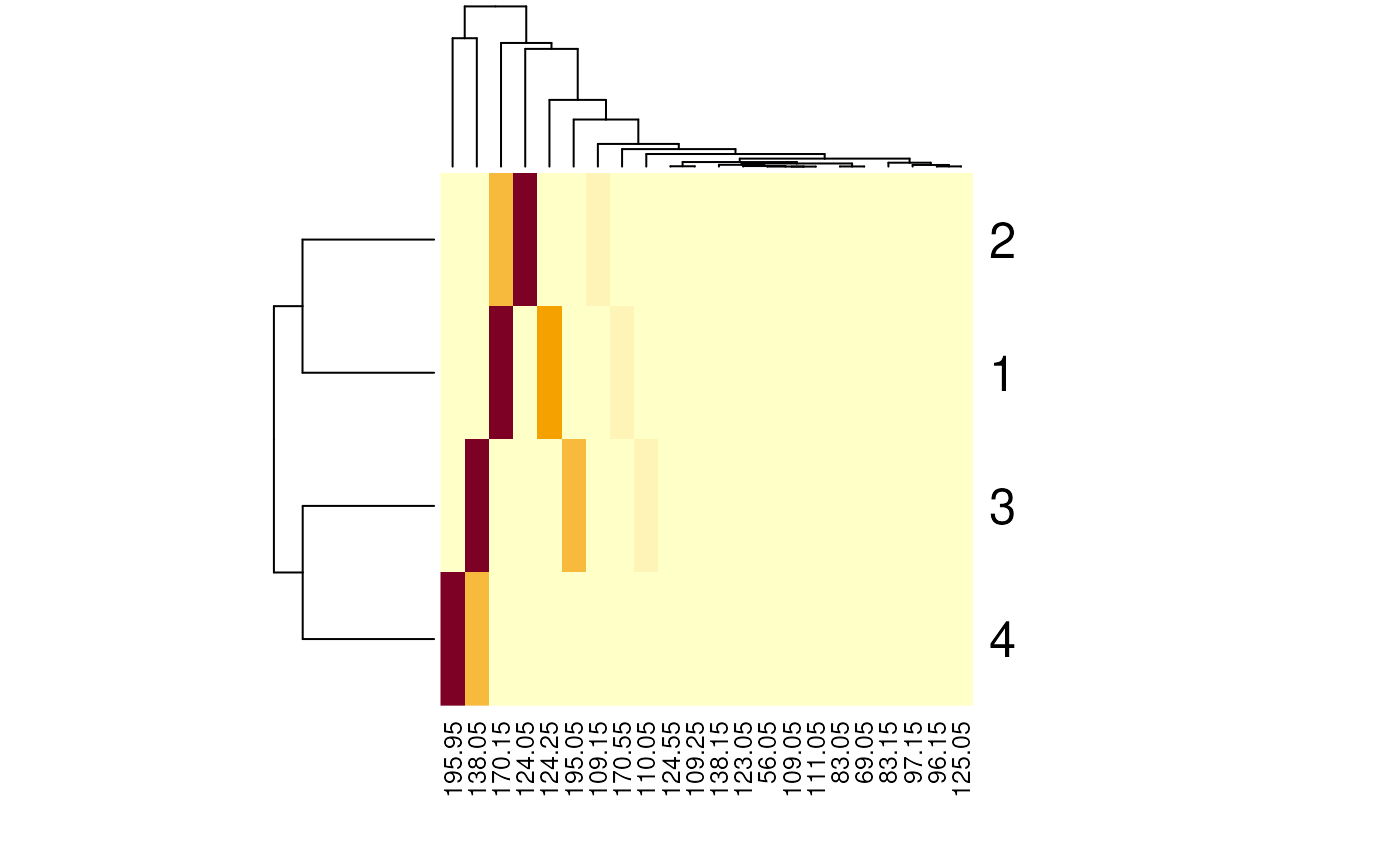
As expected, the first 2 and the last 2 spectra are more similar and are clustered together.
Exporting spectra
Spectra data can be exported with the export() method.
This method takes the Spectra that is supposed to be
exported and the backend (parameter backend) which should
be used to export the data and additional parameters for the export
function of this backend. The backend thus defines the format of the
exported file. Note however that not all MsBackend classes
might support data export. The backend classes currently supporting data
export and its format are: - MsBackendMzR
(Spectra package): export data in mzML and
mzXML format. Can not export all custom, user specified spectra
variables. - MsBackendMgf (MsBackendMgf
package): exports data in Mascot Generic Format (mgf). Exports
all spectra variables as individual spectrum fields in the mgf file. -
MsBackendMsp (MsBackendMsp):
exports data in NIST MSP format. - MsBackendMassbank (MsBackendMassbank)
exports data in Massbank text file format.
In the example below we use the MsBackendMzR to export
all spectra from the variable sps to an mzML file. We thus
pass the data, the backend that should be used for the export and the
file name of the result file (a temporary file) to the
export() function (see also the help page of the
export,MsBackendMzR function for additional supported
parameters).
fl <- tempfile()
export(sps, MsBackendMzR(), file = fl)## Writing file file18434218f081...OKTo evaluate which of the spectra variables were exported, we load the exported data again and identify spectra variables in the original file which could not be exported (because they are not defined variables in the mzML standard).
sps_im <- Spectra(backendInitialize(MsBackendMzR(), fl))
spectraVariables(sps)[!spectraVariables(sps) %in% spectraVariables(sps_im)]## [1] "id" "name" "splash" "instrument"These additional variables were thus not exported. How data export is
performed and handled depends also on the used backend. The
MsBackendMzR for example exports all spectra by default to
a single file (specified with the file parameter), but it
allows also to specify for each individual spectrum in the
Spectra to which file it should be exported (parameter
file has thus to be of length equal to the number of
spectra). As an example we export below the spectrum 1 and 3 to one file
and spectra 2 and 4 to another.
## Writing file file18439cadaf3...OK
## Writing file file184312614ca7...OKA more realistic use case for mzML export would be to export MS data
after processing, such as smoothing (using the smooth()
function) and centroiding (using the pickPeaks() function)
of raw profile-mode MS data.
Changing backends
In the previous sections we learned already that a
Spectra object can use different backends for the actual
data handling. It is also possible to change the backend of a
Spectra to a different one with the
setBackend() function. We could for example change the
(MsBackendMzR) backend of the sps_sciex object
to a MsBackendMemory backend to enable use of the data even
without the need to keep the original mzML files. Below we change the
backend of sps_sciex to the in-memory
MsBackendMemory backend.
print(object.size(sps_sciex), units = "Mb")## 0.4 Mb
sps_sciex <- setBackend(sps_sciex, MsBackendMemory())
sps_sciex## MSn data (Spectra) with 1862 spectra in a MsBackendMemory backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 1 0.280 1
## 2 1 0.559 2
## 3 1 0.838 3
## 4 1 1.117 4
## 5 1 1.396 5
## ... ... ... ...
## 1858 1 258.636 927
## 1859 1 258.915 928
## 1860 1 259.194 929
## 1861 1 259.473 930
## 1862 1 259.752 931
## ... 34 more variables/columns.
## Processing:
## Switch backend from MsBackendMzR to MsBackendMemory [Fri Jun 12 14:35:50 2026]With the call the full peak data was imported from the original mzML files into the object. This has obviously an impact on the object’s size, which is now much larger than before.
print(object.size(sps_sciex), units = "Mb")## 53.2 MbThe dataStorage spectrum variable has now changed, while
dataOrigin still keeps the information about the
originating files:
head(dataStorage(sps_sciex))## [1] "<memory>" "<memory>" "<memory>" "<memory>" "<memory>" "<memory>"
head(basename(dataOrigin(sps_sciex)))## [1] "7482fe40e87_7859" "7482fe40e87_7859" "7482fe40e87_7859" "7482fe40e87_7859"
## [5] "7482fe40e87_7859" "7482fe40e87_7859"Backends
Backends allow to use different backends to store mass
spectrometry data while providing via the Spectra
class a unified interface to use that data. This is a further
abstraction to the on-disk and in-memory data modes
from MSnbase (Gatto et al.
2020). The Spectra package defines a set of example
backends but any object extending the base MsBackend class
could be used instead. The default backends are:
MsBackendMemory: the default backend to store data in memory. Due to its design theMsBackendMemoryprovides fast access to the peaks matrices (using thepeaksData()function) and is also optimized for fast access to spectra variables and subsetting. Since all data is kept in memory, this backend has a relatively large memory footprint (depending on the data) and is thus not suggested for very large MS experiments.MsBackendDataFrame: the mass spectrometry data is stored (in-memory) in aDataFrame. Keeping the data in memory guarantees high performance but has also, depending on the number of mass peaks in each spectrum, a much higher memory footprint.MsBackendMzR: this backend keeps only general spectra variables in memory and relies on the mzR package to read mass peaks (m/z and intensity values) from the original MS files on-demand.MsBackendHdf5Peaks: similar toMsBackendMzRthis backend reads peak data only on-demand from disk while all other spectra variables are kept in memory. The peak data are stored in Hdf5 files which guarantees scalability.
All of the above mentioned backends support changing all of their
their spectra variables, except the
MsBackendMzR that does not support changing m/z or
intensity values for the mass peaks.
With the example below we load the data from a single mzML file and
use a MsBackendHdf5Peaks backend for data storage. The
hdf5path parameter allows us to specify the storage
location of the HDF5 file.
fl <- MsDataHub::TMT_Erwinia_1uLSike_Top10HCD_isol2_45stepped_60min_01.20141210.mzML.gz()## see ?MsDataHub and browseVignettes('MsDataHub') for documentation## loading from cache
sps_tmt <- Spectra(fl, backend = MsBackendHdf5Peaks(), hdf5path = tempdir())
head(basename(dataStorage(sps_tmt)))## [1] "74861e1f51f_7858.h5" "74861e1f51f_7858.h5" "74861e1f51f_7858.h5"
## [4] "74861e1f51f_7858.h5" "74861e1f51f_7858.h5" "74861e1f51f_7858.h5"A (possibly incomplete) list of R packages providing additional backends that add support for additional data types or storage options is provided below:
MsBackendCompDb(package CompoundDb): provides access to spectra data (spectra and peaks variables) from a CompDb database. Has a small memory footprint because all data (except precursor m/z values) are retrieved on-the-fly from the database.MsBackendHmdbXml(packageMsbackendHmdb): allows import of MS data from xml files of the Human Metabolome Database (HMDB). Extends theMsBackendDataFrameand keeps thus all data, after import, in memory.MsBackendMassbank(package MsBackendMassbank): allows to import/export data in MassBank text file format. Extends theMsBackendDataFrameand keeps thus all data, after import, in memory.MsBackendMassbankSql(package MsBackendMassbank): allows to directly connect to a MassBank SQL database to retrieve all MS data and variables. Has a minimal memory footprint because all data is retrieved on-the-fly from the SQL database.MsBackendMetaboLights(packager BiocStyle::Biocpkg("MsBackendMetaboLights")): retrieves and caches MS data files from the MetaboLights repository.MsBackendMgf: (package MsBackendMgf): support for import/export of mass spectrometry files in mascot generic format (MGF).MsBackendMsp: (package MsBackendMsp): allows to import/export data in NIST MSP format. Extends theMsBackendDataFrameand keeps thus all data, after import, in memory.MsBackendRawFileReader(package MsBackendRawFileReader): implements a backend for reading MS data from Thermo Fisher Scientific’s raw data files using the manufacturer’s NewRawFileReader .Net libraries. The package generalizes the functionality introduced by the rawrr package, see also (Kockmann and Panse 2021).MsBackendSql(package MsBackendSql): stores all MS data in a SQL database and has thus a minimal memory footprint.MsBackendTimsTof(packageMsBackendTimsTof: allows import of data from Bruker TimsTOF raw data files (using theopentimsrR package).MsBackendWeizMass(packageMsBackendWeizMass: allows to access MS data from WeizMass MS/MS spectral databases.
Handling very large data sets
The Spectra package was designed to support also
efficient processing of very large data sets. Most of the functionality
do not require to keep the full MS data in memory (specifically, the
peaks data, i.e., m/z and intensity values, which represent the largest
chunk of data for MS experiments). For some functions however the peaks
data needs to be loaded into memory. One such example is the
lengths() function to determine the number of peaks per
spectra that is calculated (on the fly) by evaluating the number of rows
of the peaks matrix. Backends such as the MsBackendMzR
perform by default any data processing separately (and eventually in
parallel) by data file and it should thus be safe to call any such
functions on a Spectra object with that backend. For other
backends (such as the MsBackendSql
or the MsBackendMassbankSql)
it is however advised to process the data in a chunk-wise
manner using the spectrapply() function with parameter
chunkSize. This will split the original
Spectra object into chunks of size chunkSize
and applies the function separately to each chunk. That way only data
from one chunk will eventually be loaded into memory in each iteration
enabling to process also very large Spectra objects on
computers with limited hardware resources. Instead of a
lengths(sps) call, the number of peaks per spectra could
also be determined (in a less memory demanding way) with
spectrapply(sps, lengths, chunkSize = 5000L). In that way
only peak data of 5000 spectra at a time will be loaded into memory.
Serializing (saving), moving and loading serialized
Spectra objects
Serializing and re-loading variables/objects during an analysis using
e.g. the save() and load() functions are
common in many workflows, especially if some of the tasks are
computationally intensive and take long time. Sometimes such serialized
objects might even be moved from one computer (or file system) to
another. These operations are unproblematic for Spectra
objects with in-memory backends such as the
MsBackendMemory or MsBackendDataFrame, that
keep all data in memory, would however break for on-disk
backends such as the MsBackendMzR if the file path to the
original data files is not identical. It is thus suggested (if the size
of the MS data respectively the available system memory allows it) to
change the backend for such Spectra objects to a
MsBackendMemory before serializing the object with
save(). For Spectra objects with a
MsBackendMzR an alternative option would be to eventually
update/adapt the path to the directory containing the raw (e.g. mzML)
data files: assuming these data files are available on both computers,
the path to the directory containing these can be updated with the
dataStorageBasePath<- function allowing thus to
move/copy serialized Spectra objects between computers or
file systems.
An example workflow could be:
files a.mzML, b.mzML are stored in a directory
/data/mzML/ on one computer. These get loaded as a
Spectra object with MsBackendMzR and then
serialized to a file A.RData.
Assuming this file gets now copied to another computer (where the
data is not available in a folder /data/mzML/) and loaded with
load().
load("A.RData")This Spectra object would not be valid because its
MsBackendMzR can no longer access the MS data in the
original data files. Assuming the user also copied the data files
a.mzML and b.mzML, but to a folder
/some_other_folder/, the base storage path of the object would
need to be adapted to match the directory where the data files are
available on the second computer:
dataStorageBasePath(A) <- "/some_other_folder"By pointing now the storage path to the new storage location of the
data files, the Spectra object A would also be
usable on the second computer.
Session information
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] msentropy_0.1.4 Rcpp_1.1.1-1.1 MsCoreUtils_1.25.4
## [4] MsDataHub_1.13.0 Spectra_1.23.3 BiocParallel_1.47.0
## [7] S4Vectors_0.51.3 BiocGenerics_0.59.7 generics_0.1.4
## [10] BiocStyle_2.41.0
##
## loaded via a namespace (and not attached):
## [1] KEGGREST_1.53.0 xfun_0.58 bslib_0.11.0
## [4] httr2_1.2.2 htmlwidgets_1.6.4 rhdf5_2.57.1
## [7] Biobase_2.73.1 rhdf5filters_1.25.0 vctrs_0.7.3
## [10] tools_4.6.0 curl_7.1.0 parallel_4.6.0
## [13] tibble_3.3.1 AnnotationDbi_1.75.0 RSQLite_3.53.1
## [16] cluster_2.1.8.2 blob_1.3.0 pkgconfig_2.0.3
## [19] data.table_1.18.4 dbplyr_2.5.2 desc_1.4.3
## [22] lifecycle_1.0.5 compiler_4.6.0 Biostrings_2.81.3
## [25] textshaping_1.0.5 Seqinfo_1.3.0 codetools_0.2-20
## [28] ncdf4_1.24 clue_0.3-68 htmltools_0.5.9
## [31] sass_0.4.10 yaml_2.3.12 crayon_1.5.3
## [34] pkgdown_2.2.0.9000 pillar_1.11.1 jquerylib_0.1.4
## [37] MASS_7.3-65 cachem_1.1.0 MetaboCoreUtils_1.21.1
## [40] ExperimentHub_3.3.0 AnnotationHub_4.3.0 tidyselect_1.2.1
## [43] digest_0.6.39 purrr_1.2.2 dplyr_1.2.1
## [46] bookdown_0.46 BiocVersion_3.24.0 fastmap_1.2.0
## [49] cli_3.6.6 magrittr_2.0.5 withr_3.0.2
## [52] filelock_1.0.3 rappdirs_0.3.4 bit64_4.8.2
## [55] XVector_0.53.0 httr_1.4.8 rmarkdown_2.31
## [58] bit_4.6.0 otel_0.2.0 png_0.1-9
## [61] ragg_1.5.2 memoise_2.0.1 evaluate_1.0.5
## [64] knitr_1.51 IRanges_2.47.2 BiocFileCache_3.3.0
## [67] rlang_1.2.0 glue_1.8.1 DBI_1.3.0
## [70] mzR_2.45.1 BiocManager_1.30.27 jsonlite_2.0.0
## [73] Rhdf5lib_2.1.0 R6_2.6.1 systemfonts_1.3.2
## [76] fs_2.1.0 ProtGenerics_1.45.0