Processing quantitative proteomics data with QFeatures
Laurent Gatto
Source:vignettes/Processing.Rmd
Processing.RmdAbstract
This vignette describes how to process quantitative mass spectrometry data with QFeatures: cleaning up unneeded feature variables, adding an experimental design, filtering out contaminants and reverse hits, managing missing values, log-transforming, normalising and aggregating data. This vignette is distributed under a CC BY-SA license.
Reading data as QFeatures
We are going to use a subset of the CPTAC study 6 containing
conditions A and B (Paulovich et al.
2010). The peptide-level data, as processed by MaxQuant (Cox and Mann 2008) is available in the
MsDataHub package:
x <- MsDataHub::cptac_a_b_peptides.txt() |>
read.delim()## see ?MsDataHub and browseVignettes('MsDataHub') for documentation## loading from cacheFrom the names of the columns, we see that the quantitative columns,
starting with "Intensity." (note the dot!) are at positions
56 to 61.
names(x)## [1] "Sequence" "N.term.cleavage.window"
## [3] "C.term.cleavage.window" "Amino.acid.before"
## [5] "First.amino.acid" "Second.amino.acid"
## [7] "Second.last.amino.acid" "Last.amino.acid"
## [9] "Amino.acid.after" "A.Count"
## [11] "R.Count" "N.Count"
## [13] "D.Count" "C.Count"
## [15] "Q.Count" "E.Count"
## [17] "G.Count" "H.Count"
## [19] "I.Count" "L.Count"
## [21] "K.Count" "M.Count"
## [23] "F.Count" "P.Count"
## [25] "S.Count" "T.Count"
## [27] "W.Count" "Y.Count"
## [29] "V.Count" "U.Count"
## [31] "Length" "Missed.cleavages"
## [33] "Mass" "Proteins"
## [35] "Leading.razor.protein" "Start.position"
## [37] "End.position" "Unique..Groups."
## [39] "Unique..Proteins." "Charges"
## [41] "PEP" "Score"
## [43] "Identification.type.6A_7" "Identification.type.6A_8"
## [45] "Identification.type.6A_9" "Identification.type.6B_7"
## [47] "Identification.type.6B_8" "Identification.type.6B_9"
## [49] "Experiment.6A_7" "Experiment.6A_8"
## [51] "Experiment.6A_9" "Experiment.6B_7"
## [53] "Experiment.6B_8" "Experiment.6B_9"
## [55] "Intensity" "Intensity.6A_7"
## [57] "Intensity.6A_8" "Intensity.6A_9"
## [59] "Intensity.6B_7" "Intensity.6B_8"
## [61] "Intensity.6B_9" "Reverse"
## [63] "Potential.contaminant" "id"
## [65] "Protein.group.IDs" "Mod..peptide.IDs"
## [67] "Evidence.IDs" "MS.MS.IDs"
## [69] "Best.MS.MS" "Oxidation..M..site.IDs"
## [71] "MS.MS.Count"## [1] 56 57 58 59 60 61We now read these data using the readQFeatures function.
The peptide level expression data will be imported into R as an instance
of class QFeatures named cptac with an assay
named peptides. We also use the fnames
argument to set the row-names of the peptides assay to the
peptide sequences.
library("QFeatures")
cptac <- readQFeatures(x, quantCols = i, name = "peptides", fnames = "Sequence")## Checking arguments.## Loading data as a 'SummarizedExperiment' object.## Formatting sample annotations (colData).## Formatting data as a 'QFeatures' object.## Setting assay rownames.
cptac## An instance of class QFeatures (type: bulk) with 1 set:
##
## [1] peptides: SummarizedExperiment with 11466 rows and 6 columnsEncoding the experimental design
Below we update the sample (column) annotations to encode the two groups, 6A and 6B, and the original sample numbers.
## DataFrame with 6 rows and 2 columns
## group sample
## <character> <integer>
## Intensity.6A_7 6A 7
## Intensity.6A_8 6A 8
## Intensity.6A_9 6A 9
## Intensity.6B_7 6B 7
## Intensity.6B_8 6B 8
## Intensity.6B_9 6B 9Filtering out contaminants and reverse hits
filterFeatures(cptac, ~ Reverse == "")## 'Reverse' found in 1 out of 1 assay(s).## An instance of class QFeatures (type: bulk) with 1 set:
##
## [1] peptides: SummarizedExperiment with 11436 rows and 6 columns
filterFeatures(cptac, ~ Potential.contaminant == "")## 'Potential.contaminant' found in 1 out of 1 assay(s).## An instance of class QFeatures (type: bulk) with 1 set:
##
## [1] peptides: SummarizedExperiment with 11385 rows and 6 columns
cptac <- cptac |>
filterFeatures(~ Reverse == "") |>
filterFeatures(~ Potential.contaminant == "")## 'Reverse' found in 1 out of 1 assay(s).## 'Potential.contaminant' found in 1 out of 1 assay(s).Removing up unneeded feature variables
The spreadsheet that was read above contained numerous variables that are returned by MaxQuant, but not necessarily necessary in the frame of a downstream statistical analysis.
rowDataNames(cptac)## CharacterList of length 1
## [["peptides"]] Sequence N.term.cleavage.window ... MS.MS.CountThe only ones that we will be needing below are the peptides
sequences and the protein identifiers. Below, we store these variables
of interest and filter them using the selectRowData
function.
rowvars <- c("Sequence", "Proteins", "Leading.razor.protein")
cptac <- selectRowData(cptac, rowvars)
rowDataNames(cptac)## CharacterList of length 1
## [["peptides"]] Sequence Proteins Leading.razor.proteinManaging missing values
Missing values can be very numerous in certain proteomics experiments
and need to be dealt with carefully. The first step is to assess their
presence across samples and features. But before being able to do so, we
need to replace 0 by NA, given that MaxQuant encodes
missing data with a 0 using the zeroIsNA function.
## $nNA
## DataFrame with 1 row and 3 columns
## assay nNA pNA
## <character> <integer> <numeric>
## 1 peptides 30609 0.449194
##
## $nNArows
## DataFrame with 11357 rows and 4 columns
## assay name nNA pNA
## <character> <character> <integer> <numeric>
## 1 peptides AAAAGAGGAG... 4 0.666667
## 2 peptides AAAALAGGK 0 0.000000
## 3 peptides AAAALAGGKK 0 0.000000
## 4 peptides AAADALSDLE... 0 0.000000
## 5 peptides AAADALSDLE... 0 0.000000
## ... ... ... ... ...
## 11353 peptides YYSIYDLGNN... 6 1.000000
## 11354 peptides YYTFNGPNYN... 3 0.500000
## 11355 peptides YYTITEVATR 4 0.666667
## 11356 peptides YYTVFDRDNN... 6 1.000000
## 11357 peptides YYTVFDRDNN... 6 1.000000
##
## $nNAcols
## DataFrame with 6 rows and 4 columns
## assay name nNA pNA
## <character> <character> <integer> <numeric>
## 1 peptides Intensity.... 4669 0.411112
## 2 peptides Intensity.... 5388 0.474421
## 3 peptides Intensity.... 5224 0.459981
## 4 peptides Intensity.... 4651 0.409527
## 5 peptides Intensity.... 5470 0.481641
## 6 peptides Intensity.... 5207 0.458484The output of the nNA function tells us that
- there are currently close to 50% is missing values in the data;
- there are 4051 peptides with 0 missing values, 989 with a single missing values, … and 3014 peptides composed of only missing values;
- the range of missing values in the 6 samples is comparable and ranges between 4651 and 5470.
In this dataset, we have such a high number of peptides without any
data because the 6 samples are a subset of a larger dataset, and these
peptides happened to be absent in groups A and B. Below, we use
filterNA to remove all the peptides that contain one or
more missing values by using pNA = 0 (which also is the
default value).
## An instance of class QFeatures (type: bulk) with 1 set:
##
## [1] peptides: SummarizedExperiment with 4051 rows and 6 columnsI we wanted to keep peptides that have up to 90% of missing values,
corresponsing in this case to those that have only one value (i.e 5/6
percent of missing values), we could have set pNA to
0.9.
Counting unique features
Counting the number of unique features across samples can be used for
quality control or for assessing the identification efficiency between
different conditions or experimental set-ups.
countUniqueFeatures can be used to count the number of
features that are contained in each sample of an assay from a
QFeatures object. For instance, we can count the number of
(non-missing) peptides per sample from the peptides assay.
Note that the counts are automatically stored in the
colData of cptac, under
peptide_counts:
cptac <- countUniqueFeatures(cptac,
i = "peptides",
colDataName = "peptide_counts")
colData(cptac)## DataFrame with 6 rows and 3 columns
## group sample peptide_counts
## <character> <integer> <integer>
## Intensity.6A_7 6A 7 4051
## Intensity.6A_8 6A 8 4051
## Intensity.6A_9 6A 9 4051
## Intensity.6B_7 6B 7 4051
## Intensity.6B_8 6B 8 4051
## Intensity.6B_9 6B 9 4051We can also count the number of unique proteins. We therefore need to
tell countUniqueFeatures that we need to group by protein
(the protein name is stored in the rowData under
Proteins):
cptac <- countUniqueFeatures(cptac,
i = "peptides",
groupBy = "Proteins",
colDataName = "protein_counts")
colData(cptac)## DataFrame with 6 rows and 4 columns
## group sample peptide_counts protein_counts
## <character> <integer> <integer> <integer>
## Intensity.6A_7 6A 7 4051 1125
## Intensity.6A_8 6A 8 4051 1125
## Intensity.6A_9 6A 9 4051 1125
## Intensity.6B_7 6B 7 4051 1125
## Intensity.6B_8 6B 8 4051 1125
## Intensity.6B_9 6B 9 4051 1125Imputation
The impute method can be used to perform missing value
imputation using a variety of imputation methods. The method takes an
instance of class QFeatures (or a
SummarizedExperiment) as input, an a character naming the
desired method (see ?impute for the complete list with
details) and returns a new instance of class QFeatures (or
SummarizedExperiment) with imputed data.
As described in more details in (Lazar et al. 2016), there are two types of mechanisms resulting in missing values in LC/MSMS experiments.
Missing values resulting from absence of detection of a feature, despite ions being present at detectable concentrations. For example in the case of ion suppression or as a result from the stochastic, data-dependent nature of the MS acquisition method. These missing value are expected to be randomly distributed in the data and are defined as missing at random (MAR) or missing completely at random (MCAR).
Biologically relevant missing values, resulting from the absence of the low abundance of ions (below the limit of detection of the instrument). These missing values are not expected to be randomly distributed in the data and are defined as missing not at random (MNAR).
MAR and MCAR values can be reasonably well tackled by many imputation methods. MNAR data, however, requires some knowledge about the underlying mechanism that generates the missing data, to be able to attempt data imputation. MNAR features should ideally be imputed with a left-censor (for example using a deterministic or probabilistic minimum value) method. Conversely, it is recommended to use hot deck methods (for example nearest neighbour, maximum likelihood, etc) when data are missing at random.
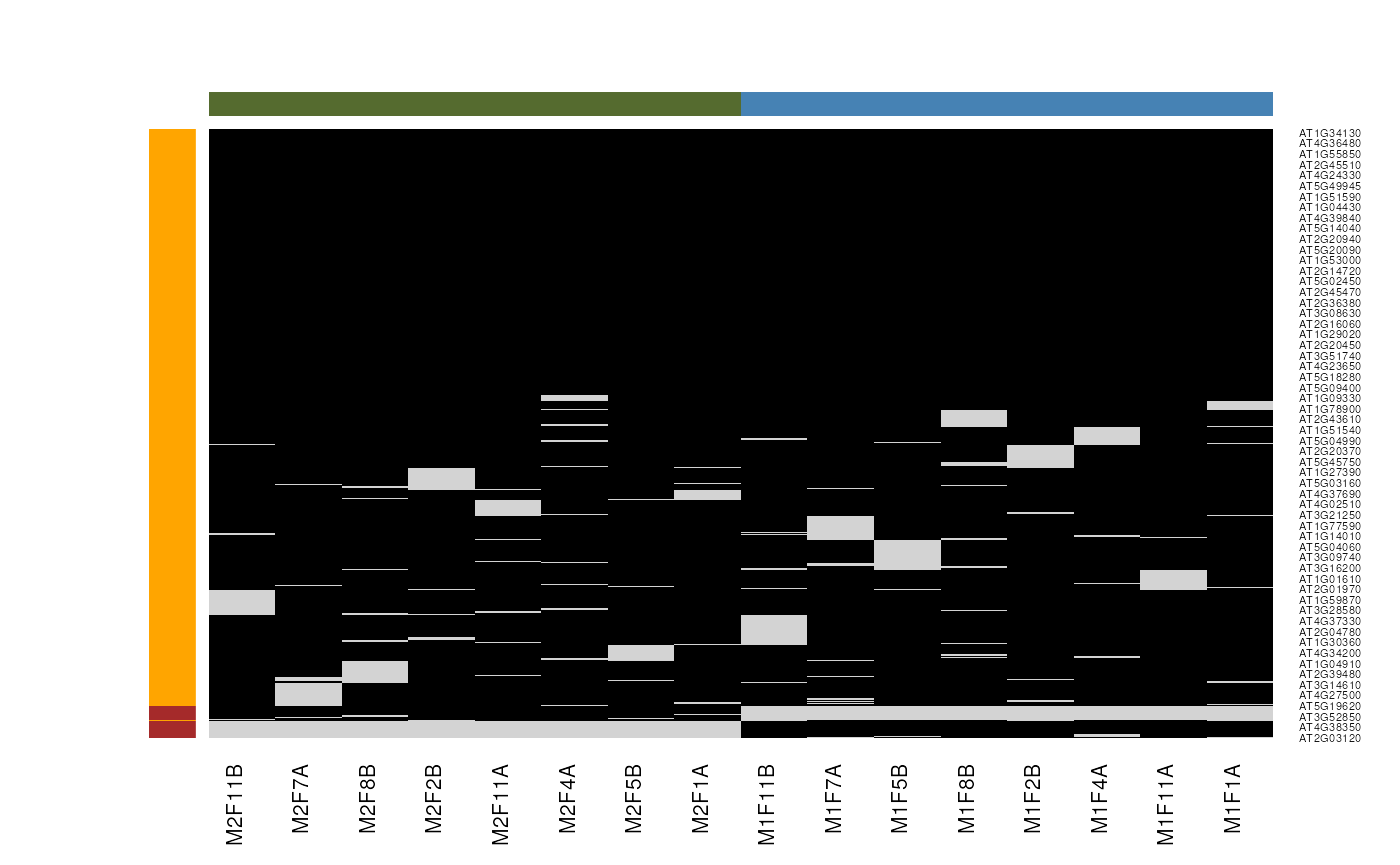
Mixed imputation method. Black cells represent presence of quantitation values and light grey corresponds to missing data. The two groups of interest are depicted in green and blue along the heatmap columns. Two classes of proteins are annotated on the left: yellow are proteins with randomly occurring missing values (if any) while proteins in brown are candidates for non-random missing value imputation.
It is anticipated that the identification of both classes of missing values will depend on various factors, such as feature intensities and experimental design. Below, we use perform mixed imputation, applying nearest neighbour imputation on the 654 features that are assumed to contain randomly distributed missing values (if any) (yellow on figure @ref(fig:miximp)) and a deterministic minimum value imputation on the 35 proteins that display a non-random pattern of missing values (brown on figure @ref(fig:miximp)).
See the dedicated Imputation
vignette, (availabel from within R with
vignette("Imputation", package = "QFeatures")) for
technical details on the imputation functions and methods.
Data transformation
When analysing continuous data using parametric methods (such as t-test or linear models), it is often necessary to log-transform the data. The figure below (left) show that how our data is mainly composed of small values with a long tail of larger ones, which is a typical pattern of quantitative omics data.
Below, we use the logTransform function to
log2-transform our data. This time, instead of overwriting the peptides
assay, we are going to create a new one to contain the log2-transformed
data.
addAssay(cptac,
logTransform(cptac[[1]]),
name = "peptides_log")The addAssay() function is the general function that
adds new assays to a QFeatures object. The step above could
more easily be exectuted with the logTransform() method,
that directly returns an updated QFeatures object. Using
logTransform() also automatically adds links between
assays.
cptac <- logTransform(cptac,
i = "peptides",
name = "peptides_log")
cptac## An instance of class QFeatures (type: bulk) with 2 sets:
##
## [1] peptides: SummarizedExperiment with 4051 rows and 6 columns
## [2] peptides_log: SummarizedExperiment with 4051 rows and 6 columns
par(mfrow = c(1, 2))
limma::plotDensities(assay(cptac[[1]]))
limma::plotDensities(assay(cptac[[2]]))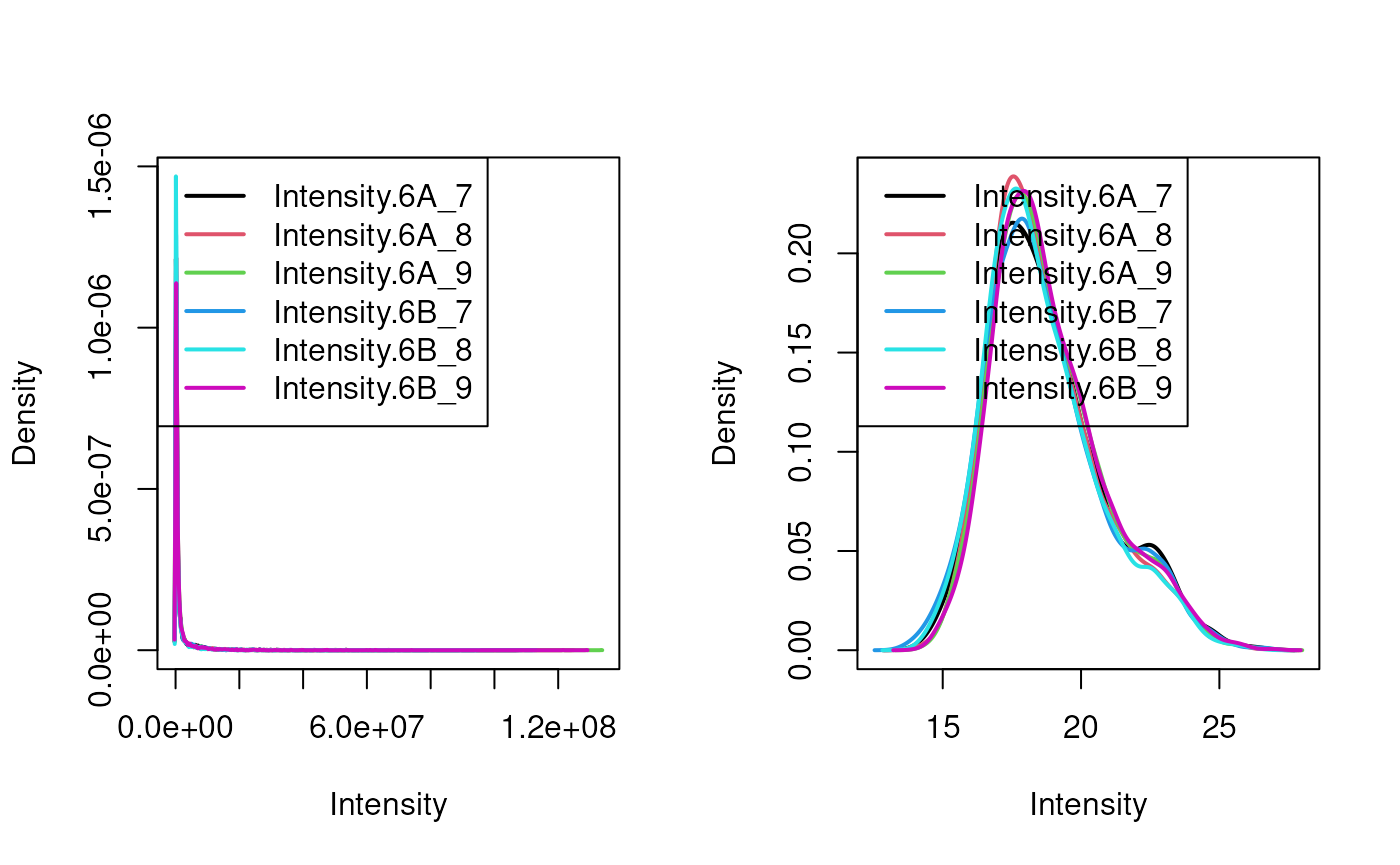
Quantitative data in its original scale (left) and log2-transformed (right).
Normalisation
Assays in QFeatures objects can be normalised with the
normalize function. The type of normalisation is defined by
the method argument; below, we use median normalisation,
store the normalised data into a new experiment, and visualise the
resulting data.
The normalize() function can also be directly applied to
the QFeatures object.
cptac <- normalize(cptac,
i = "peptides_log",
name = "peptides_norm",
method = "diff.median")
cptac## An instance of class QFeatures (type: bulk) with 3 sets:
##
## [1] peptides: SummarizedExperiment with 4051 rows and 6 columns
## [2] peptides_log: SummarizedExperiment with 4051 rows and 6 columns
## [3] peptides_norm: SummarizedExperiment with 4051 rows and 6 columnsIt is also possible to extract and normalise the
peptides_log SummarizedExperiment and add it
back to the QFeatures object with
addAssay().
addAssay(cptac,
normalize(cptac[["peptides_log"]],
method = "center.median"),
name = "peptides_norm")
par(mfrow = c(1, 2))
limma::plotDensities(assay(cptac[["peptides_log"]]))
limma::plotDensities(assay(cptac[["peptides_norm"]]))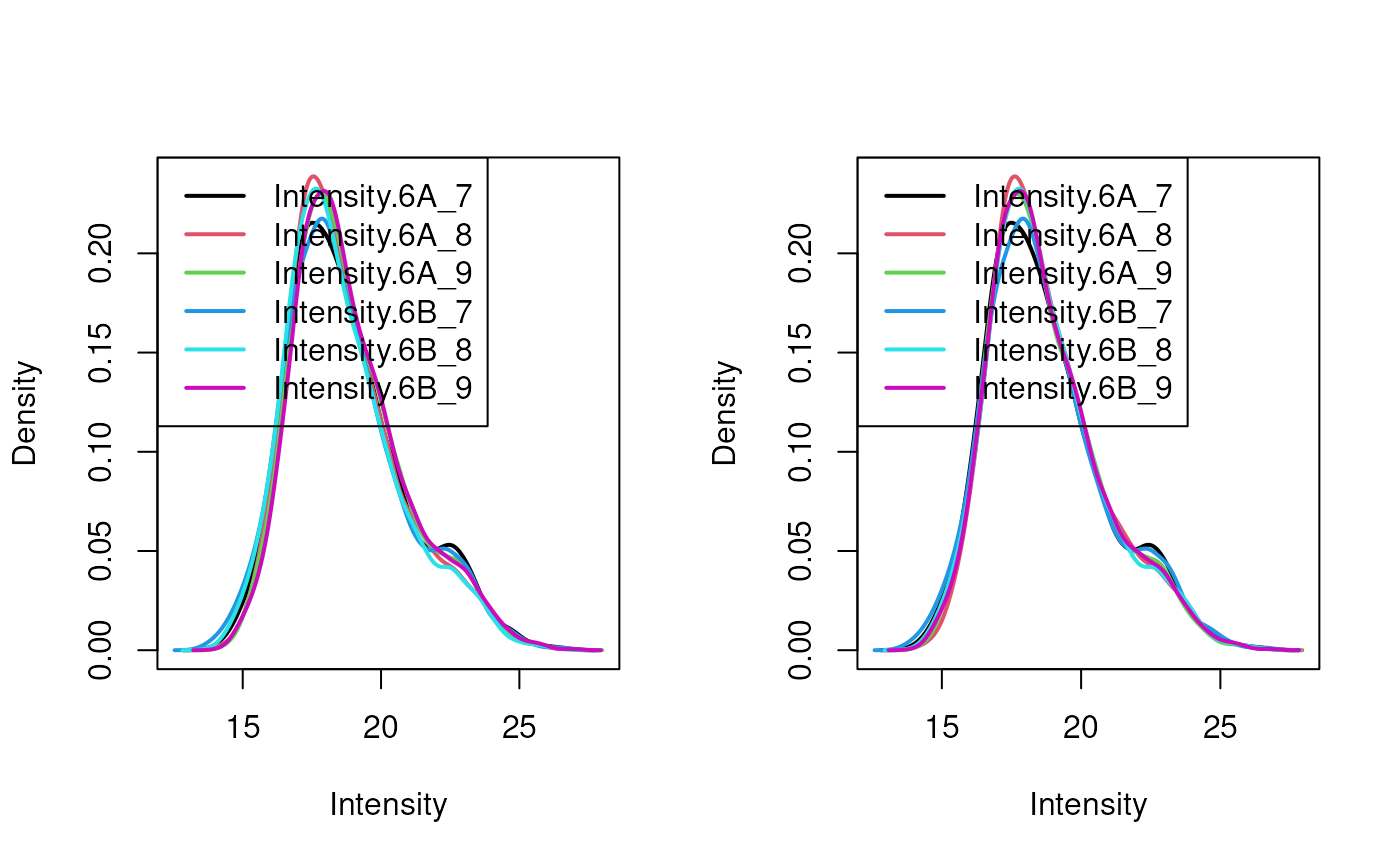
Distribution of log2 peptide intensities before (left) and after (right) median normalisation.
Feature aggregation
At this stage, it is possible to directly use the peptide-level intensities to perform a statistical analysis (Goeminne et al. 2016), or aggregate the peptide-level data into protein intensities, and perform the differential expression analysis at the protein level.
To aggregate feature data, we can use the
aggregateFeatures function that takes the following
inputs:
- the name of the
QFeaturesinstance that contains the peptide quantitation data -"cptac"in our example; -
i: the name or index of the assay that contains the (normalised) peptide quantitation data -"peptides_norm"in our case; -
fcol: the feature variable (in the assay above) to be used to define what peptides to aggregate -"Proteins"here, given that we want to aggregate all peptides that belong to one protein (group); -
name: the name of the new aggregates assay -"proteins"in this case; - and finally
fun, the function that will compute this aggregation - we will be using the default value, namelyrobustSummary(Sticker et al. 2019).
cptac <- aggregateFeatures(cptac,
i = "peptides_norm",
fcol = "Proteins",
name = "proteins")## | | | 0%## | |======================================================================| 100%
cptac## An instance of class QFeatures (type: bulk) with 4 sets:
##
## [1] peptides: SummarizedExperiment with 4051 rows and 6 columns
## [2] peptides_log: SummarizedExperiment with 4051 rows and 6 columns
## [3] peptides_norm: SummarizedExperiment with 4051 rows and 6 columns
## [4] proteins: SummarizedExperiment with 1125 rows and 6 columnsWe obtain a final 1125 quantified proteins in the new
proteins assay. Below, we display the quantitation data for
the first 6 proteins and their respective variables. The latter shown
that number of peptides that were using during the aggregation step
(.n column).
head(assay(cptac[["proteins"]]))## Intensity.6A_7 Intensity.6A_8
## P00918ups|CAH2_HUMAN_UPS 17.23988 16.98222
## P01008ups|ANT3_HUMAN_UPS;CON__P41361 16.81917 16.11327
## P01127ups|PDGFB_HUMAN_UPS 16.45163 16.90199
## P02144ups|MYG_HUMAN_UPS 16.81662 16.55897
## P02753ups|RETBP_HUMAN_UPS 17.80433 16.79555
## P02787ups|TRFE_HUMAN_UPS 16.74488 16.97394
## Intensity.6A_9 Intensity.6B_7
## P00918ups|CAH2_HUMAN_UPS 16.63167 18.27738
## P01008ups|ANT3_HUMAN_UPS;CON__P41361 16.33382 16.72030
## P01127ups|PDGFB_HUMAN_UPS 16.83464 18.19830
## P02144ups|MYG_HUMAN_UPS 17.28038 17.86570
## P02753ups|RETBP_HUMAN_UPS 16.55527 18.39382
## P02787ups|TRFE_HUMAN_UPS 16.34641 18.13812
## Intensity.6B_8 Intensity.6B_9
## P00918ups|CAH2_HUMAN_UPS 18.54886 18.46754
## P01008ups|ANT3_HUMAN_UPS;CON__P41361 16.74130 16.48097
## P01127ups|PDGFB_HUMAN_UPS 18.77132 17.16724
## P02144ups|MYG_HUMAN_UPS 18.55434 18.29205
## P02753ups|RETBP_HUMAN_UPS 17.73507 18.15238
## P02787ups|TRFE_HUMAN_UPS 18.51059 18.15718
rowData(cptac[["proteins"]])## DataFrame with 1125 rows and 3 columns
## Proteins Leading.razor.protein
## <character> <character>
## P00918ups|CAH2_HUMAN_UPS P00918ups|... P00918ups|...
## P01008ups|ANT3_HUMAN_UPS;CON__P41361 P01008ups|... P01008ups|...
## P01127ups|PDGFB_HUMAN_UPS P01127ups|... P01127ups|...
## P02144ups|MYG_HUMAN_UPS P02144ups|... P02144ups|...
## P02753ups|RETBP_HUMAN_UPS P02753ups|... P02753ups|...
## ... ... ...
## sp|Q99207|NOP14_YEAST sp|Q99207|... sp|Q99207|...
## sp|Q99216|PNO1_YEAST sp|Q99216|... sp|Q99216|...
## sp|Q99257|MEX67_YEAST sp|Q99257|... sp|Q99257|...
## sp|Q99258|RIB3_YEAST sp|Q99258|... sp|Q99258|...
## sp|Q99383|HRP1_YEAST sp|Q99383|... sp|Q99383|...
## .n
## <integer>
## P00918ups|CAH2_HUMAN_UPS 1
## P01008ups|ANT3_HUMAN_UPS;CON__P41361 1
## P01127ups|PDGFB_HUMAN_UPS 1
## P02144ups|MYG_HUMAN_UPS 1
## P02753ups|RETBP_HUMAN_UPS 2
## ... ...
## sp|Q99207|NOP14_YEAST 1
## sp|Q99216|PNO1_YEAST 1
## sp|Q99257|MEX67_YEAST 2
## sp|Q99258|RIB3_YEAST 2
## sp|Q99383|HRP1_YEAST 2We can get a quick overview of this .n variable by
computing the table below, that shows us that we have 405 proteins that
are based on a single peptides, 230 that are based on two, 119 that are
based on three, … and a single protein that is the results of
aggregating 44 peptides.
table(rowData(cptac[["proteins"]])$.n)##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
## 405 230 119 84 64 53 37 29 24 24 13 9 4 3 3 7 3 1 1 1
## 21 22 23 24 25 30 31 33 44
## 1 2 2 1 1 1 1 1 1Let’s choose P02787ups|TRFE_HUMAN_UPS and visualise its
expression pattern in the 2 groups at the protein and (log-tranformed
and normalised) peptide level. We drop the first peptide-level assay as
it is on a different scale (i.e. not log-transformed).
library("ggplot2")
library("dplyr")
longForm(cptac["P02787ups|TRFE_HUMAN_UPS", , -1]) |>
as.data.frame() |>
mutate(group = ifelse(grepl("A", colname), "A", "B")) |>
mutate(sample = sub("Intensity\\.", "", colname)) |>
ggplot(aes(x = sample, y = value,
colour = rowname,
shape = group)) +
geom_line(aes(group = rowname)) +
geom_point(size = 3) +
facet_grid(~ assay)## Warning: 'experiments' dropped; see 'drops()'## harmonizing input:
## removing 6 sampleMap rows not in names(experiments)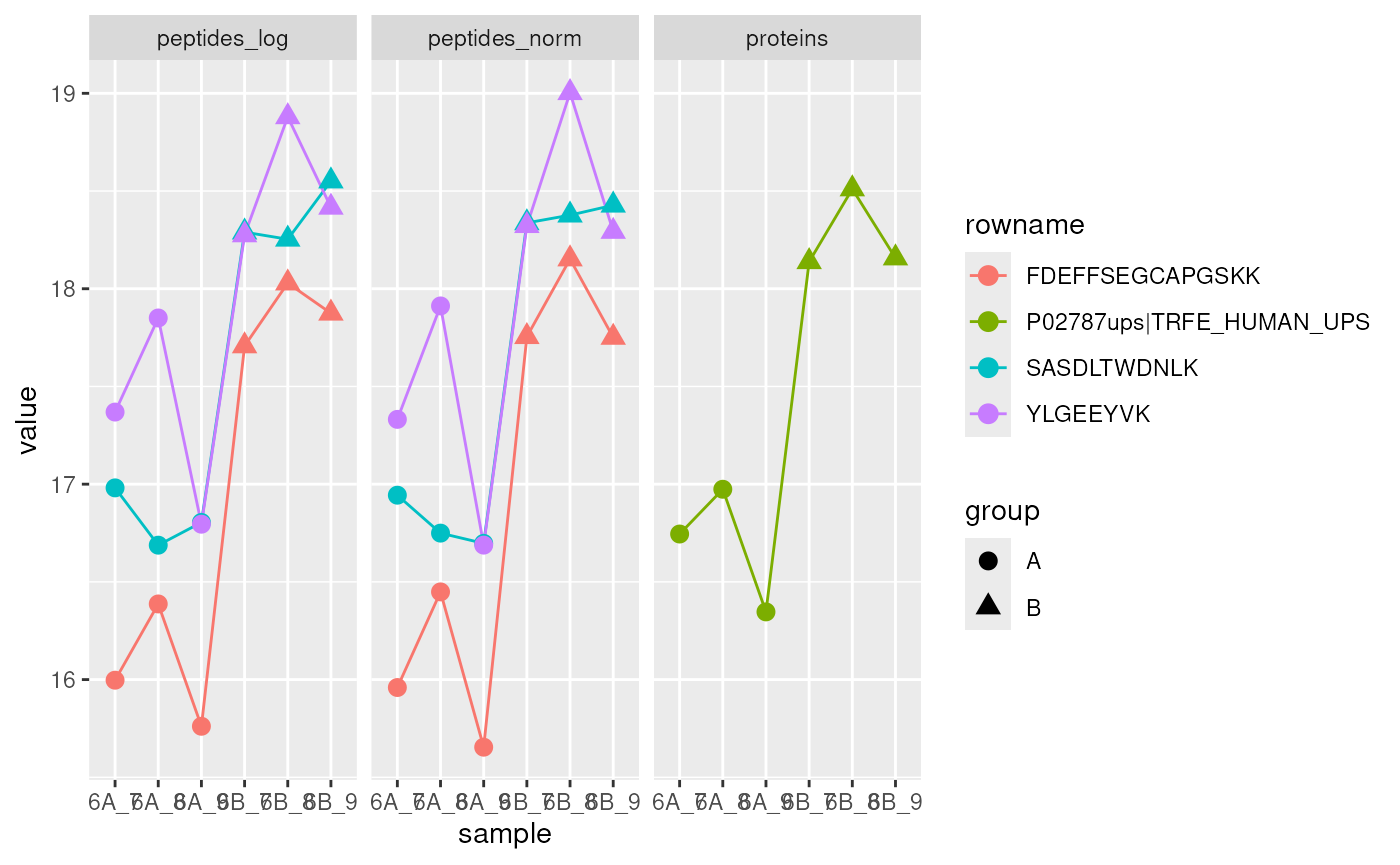
Expression intensities for the protein P02787ups|TRFE_HUMAN_UPS (right, green) and its peptides (left) in groups A (circles) and B (triangles).
See also
- The other vignettes in the
QFeaturespackage. - The QFeaturesWorkshop2020 workshop, presented at the EuroBioc2020 meeting. It also documents how to use a custom docker container to run the workshop code.
- The Quantitative proteomics data analysis chapter of the WSBIM2122 course.
Session information
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] gplots_3.3.0 MsDataHub_1.12.0
## [3] dplyr_1.2.1 ggplot2_4.0.3
## [5] QFeatures_1.23.1 MultiAssayExperiment_1.38.0
## [7] SummarizedExperiment_1.42.0 Biobase_2.72.0
## [9] GenomicRanges_1.64.0 Seqinfo_1.2.0
## [11] IRanges_2.46.0 S4Vectors_0.50.0
## [13] BiocGenerics_0.58.0 generics_0.1.4
## [15] MatrixGenerics_1.24.0 matrixStats_1.5.0
## [17] BiocStyle_2.40.0
##
## loaded via a namespace (and not attached):
## [1] bitops_1.0-9 DBI_1.3.0 httr2_1.2.2
## [4] rlang_1.2.0 magrittr_2.0.5 clue_0.3-68
## [7] otel_0.2.0 compiler_4.6.0 RSQLite_2.4.6
## [10] png_0.1-9 systemfonts_1.3.2 vctrs_0.7.3
## [13] reshape2_1.4.5 stringr_1.6.0 ProtGenerics_1.44.0
## [16] pkgconfig_2.0.3 crayon_1.5.3 fastmap_1.2.0
## [19] dbplyr_2.5.2 XVector_0.52.0 labeling_0.4.3
## [22] caTools_1.18.3 rmarkdown_2.31 ragg_1.5.2
## [25] purrr_1.2.2 bit_4.6.0 xfun_0.57
## [28] cachem_1.1.0 jsonlite_2.0.0 blob_1.3.0
## [31] DelayedArray_0.38.1 cluster_2.1.8.2 R6_2.6.1
## [34] bslib_0.10.0 stringi_1.8.7 RColorBrewer_1.1-3
## [37] limma_3.68.0 jquerylib_0.1.4 Rcpp_1.1.1-1.1
## [40] bookdown_0.46 knitr_1.51 BiocBaseUtils_1.14.0
## [43] Matrix_1.7-5 igraph_2.3.0 tidyselect_1.2.1
## [46] abind_1.4-8 yaml_2.3.12 curl_7.1.0
## [49] lattice_0.22-9 tibble_3.3.1 plyr_1.8.9
## [52] withr_3.0.2 KEGGREST_1.52.0 S7_0.2.2
## [55] evaluate_1.0.5 desc_1.4.3 BiocFileCache_3.2.0
## [58] ExperimentHub_3.2.0 Biostrings_2.80.0 pillar_1.11.1
## [61] BiocManager_1.30.27 filelock_1.0.3 KernSmooth_2.23-26
## [64] BiocVersion_3.23.1 scales_1.4.0 gtools_3.9.5
## [67] glue_1.8.1 lazyeval_0.2.3 tools_4.6.0
## [70] AnnotationHub_4.2.0 fs_2.1.0 grid_4.6.0
## [73] tidyr_1.3.2 MsCoreUtils_1.25.3 AnnotationDbi_1.74.0
## [76] cli_3.6.6 rappdirs_0.3.4 textshaping_1.0.5
## [79] S4Arrays_1.12.0 AnnotationFilter_1.36.0 gtable_0.3.6
## [82] sass_0.4.10 digest_0.6.39 SparseArray_1.12.2
## [85] htmlwidgets_1.6.4 farver_2.1.2 memoise_2.0.1
## [88] htmltools_0.5.9 pkgdown_2.2.0.9000 lifecycle_1.0.5
## [91] httr_1.4.8 statmod_1.5.1 bit64_4.8.0
## [94] MASS_7.3-65License
This vignette is distributed under a CC BY-SA license license.