Load mass spectrometry-based proteomics data using `readQFeatures()`
Laurent Gatto
Christophe Vanderaa
14 April 2026
Source:vignettes/read_QFeatures.Rmd
read_QFeatures.RmdThe QFeatures class
The QFeatures class stores data as a list of
SummarizedExperiment objects that contain data processed at
different levels. For instance, a QFeatures object may
contain data at the peptide-to-spectrum-match (PSM) level, at the
peptide level and at the protein level. We call each
SummarizedExperiment object contained in a
QFeatures object a set. Because the different sets
are often related, they often share the same samples (columns).
QFeatures automatically creates links between the related
samples and their annotations (stored in a single colData
table). Similarly, different sets often share related features (rows).
For instance, proteins are composed of peptides and peptides are
composed of PSMs. QFeatures automatically creates links
between the related features through an AssayLinks
object.
) on `SingleCellExperiment` and `QFeatures` objects](figs/readQFeatures_class.png)
The QFeatures data class. The QFeatures object
contains a list of SummarizedExperiment ojects (see class
description) on SingleCellExperiment and
QFeatures objects
Converting tabular data
QFeatures is designed to process and manipulate the
MS-based proteomics data obtained after identification and
quantification of the raw MS files. The identification and
quantification steps are generally performed by dedicated software
(e.g. Sage, FragPipe, Proteome Discoverer, MaxQuant, …) that return a
set of tabular data. readQFeatures() converts these tabular
data into a QFeatures object. We refer to these tables as
the assayData tables.
We distinguish between two use cases: the single-set case and the multi-set case.
The single-set case
The single-set case will generate a QFeatures object
with a single SummarizedExperiment object. This is
generally the case when reading data at the peptide or protein level, or
when the samples where multiplexed (e.g. using TMT) within a single MS
run. There are two types of columns:
- Quantitative columns (
quantCols): 1 to n (depending on technology) - Feature annotations: e.g. peptide sequence, ion charge, protein name
In this case, each quantitative column contains information for a single sample. This can be schematically represented as below:
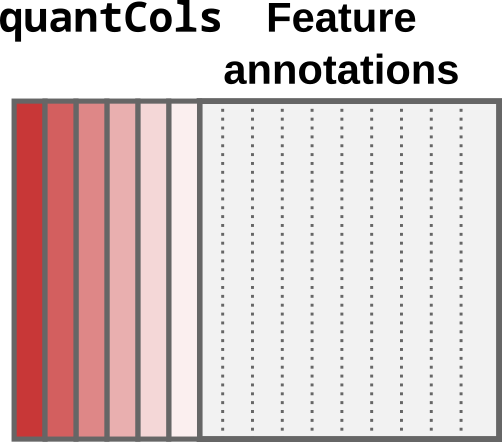
Schematic representation of a data table under the single-set case.
Quantification columns (quantCols) are represented by
different shades of red.
The hyperLOPIT data is an example data that falls under the
single-set case (see ?hlpsms for more details). The
quantCols are X126, X127N,
X127C, …, X130N, X130C,
X131 and correspond to different TMT labels.
In this toy example, there are 3,010 rows corresponding to features
(quantified PSMs) and 28 columns corresponding to different data fields
generated by MaxQuant during the analysis of the raw MS spectra. The
table is converted to a QFeatures object as follows:
data("hlpsms")
quantCols <- grep("^X", colnames(hlpsms))
(qfSingle <- readQFeatures(hlpsms, quantCols = quantCols))
#> Checking arguments.
#> Loading data as a 'SummarizedExperiment' object.
#> Formatting sample annotations (colData).
#> Formatting data as a 'QFeatures' object.
#> An instance of class QFeatures (type: bulk) with 1 set:
#>
#> [1] quants: SummarizedExperiment with 3010 rows and 10 columnsThe object returned by readQFeatures() is a
QFeatures object containing 1
SummarizedExperiment set. The set is named
quants by default, but we could name it psms
by providing the name argument:
(qfSingle <- readQFeatures(hlpsms, quantCols = quantCols, name = "psms"))
#> Checking arguments.
#> Loading data as a 'SummarizedExperiment' object.
#> Formatting sample annotations (colData).
#> Formatting data as a 'QFeatures' object.
#> An instance of class QFeatures (type: bulk) with 1 set:
#>
#> [1] psms: SummarizedExperiment with 3010 rows and 10 columnsThe multi-set case
The multi-set case will generate a QFeatures object with
multiple SummarizedExperiment objects. This is generally
the case when reading data at the PSM level that has been acquired as
part of multiple runs. In this case, the identification and
quantification software concatenates the results across MS runs in a
single table. There are three types of columns:
- Run identifier column (
runCol): e.g. file name. - Quantification columns (
quantCols): 1 to n (depending on technology). - Feature annotations: e.g. peptide sequence, ion charge, protein name.
Each quantitative column contains information for multiple samples. This can be schematically represented as below:
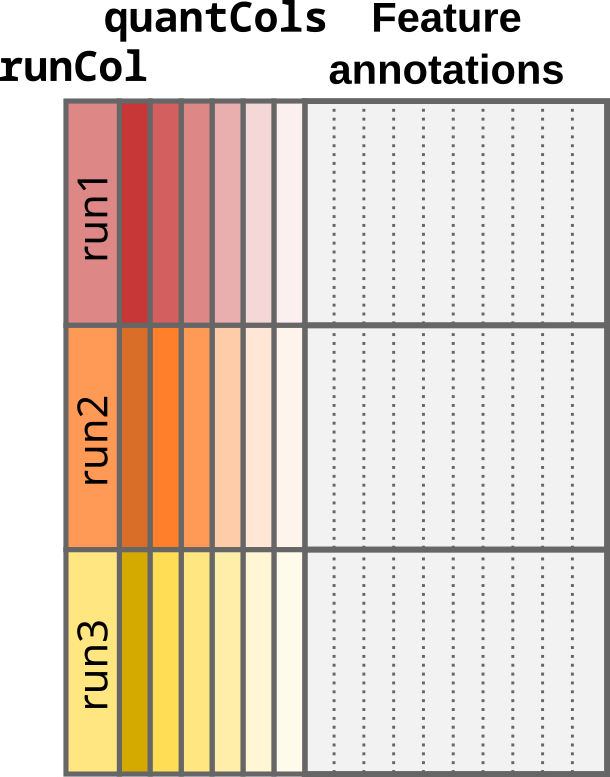
Schematic representation of a data table under the multi-set case.
Quantification columns (quantCols) are coloured by run and
shaded by label. Every sample is uniquely represented by a colour and
shade. Note that every quantCol contains multiple samples.
We will again use hyperLOPIT data and simulate it was acquired as part of multiple runs, hence falling under the multi-set case. The MS run is often identified with the name of the file it generated.
Note that the data set now has a column called “FileName” with 3 different runs:
To avoid that a quantification column contains data from multiple
samples, readQFeatures() splits the table into mulitple set
depending on the runCol column, here given as
FileName:
(qfMulti <- readQFeatures(hlpsms, quantCols = quantCols, runCol = "FileName"))
#> Checking arguments.
#> Loading data as a 'SummarizedExperiment' object.
#> Splitting data in runs.
#> | | | 0% | |======================= | 33% | |=============================================== | 67% | |======================================================================| 100%
#> Formatting sample annotations (colData).
#> Formatting data as a 'QFeatures' object.
#> An instance of class QFeatures (type: bulk) with 3 sets:
#>
#> [1] run1.raw: SummarizedExperiment with 1004 rows and 10 columns
#> [2] run2.raw: SummarizedExperiment with 1004 rows and 10 columns
#> [3] run3.raw: SummarizedExperiment with 1002 rows and 10 columnsThe object returned by readQFeatures() is a
QFeatures object containing 3
SummarizedExperiment sets. The sets are automatically named
based on the values found in runCol.
Including sample annotations
Data often comes with sample annotations that provide information
about the experimental design. These data are generally created by the
user. To facilitate sample annotations, readQFeatures()
also allows providing the annotation table as the colData
argument. Depending on the use case, one or multiple columns are
required.
For the single-set case, the colData table must contain
a column named quantCols.

colData for the single-set case
Let’s simulate such a table:
(coldata <- DataFrame(
quantCols = quantCols,
condition = rep(c("A", "B"), 5),
batch = rep(c("batch1", "batch2"), each = 5)
))
#> DataFrame with 10 rows and 3 columns
#> quantCols condition batch
#> <integer> <character> <character>
#> 1 1 A batch1
#> 2 2 B batch1
#> 3 3 A batch1
#> 4 4 B batch1
#> 5 5 A batch1
#> 6 6 B batch2
#> 7 7 A batch2
#> 8 8 B batch2
#> 9 9 A batch2
#> 10 10 B batch2We can now provide the table to readQFeatures():
(qfSingle <- readQFeatures(hlpsms, quantCols = quantCols, colData = coldata))
#> Checking arguments.
#> Loading data as a 'SummarizedExperiment' object.
#> Formatting sample annotations (colData).
#> Formatting data as a 'QFeatures' object.
#> An instance of class QFeatures (type: bulk) with 1 set:
#>
#> [1] quants: SummarizedExperiment with 3010 rows and 10 columnsFor convenience, the quantCols argument can be omitted
when providing colData (quantCols are then
fetched from this table):
(qfSingle <- readQFeatures(hlpsms, colData = coldata))
#> Checking arguments.
#> Loading data as a 'SummarizedExperiment' object.
#> Formatting sample annotations (colData).
#> Formatting data as a 'QFeatures' object.
#> An instance of class QFeatures (type: bulk) with 1 set:
#>
#> [1] quants: SummarizedExperiment with 3010 rows and 10 columnsThe annotations are retrieved as follows:
colData(qfSingle)
#> DataFrame with 10 rows and 3 columns
#> quantCols condition batch
#> <integer> <character> <character>
#> X126 NA NA NA
#> X127C NA NA NA
#> X127N NA NA NA
#> X128C NA NA NA
#> X128N NA NA NA
#> X129C NA NA NA
#> X129N NA NA NA
#> X130C NA NA NA
#> X130N NA NA NA
#> X131 NA NA NAFor the multi-set case, the colData table must contain a
column named quantCols and a column called
runCol.
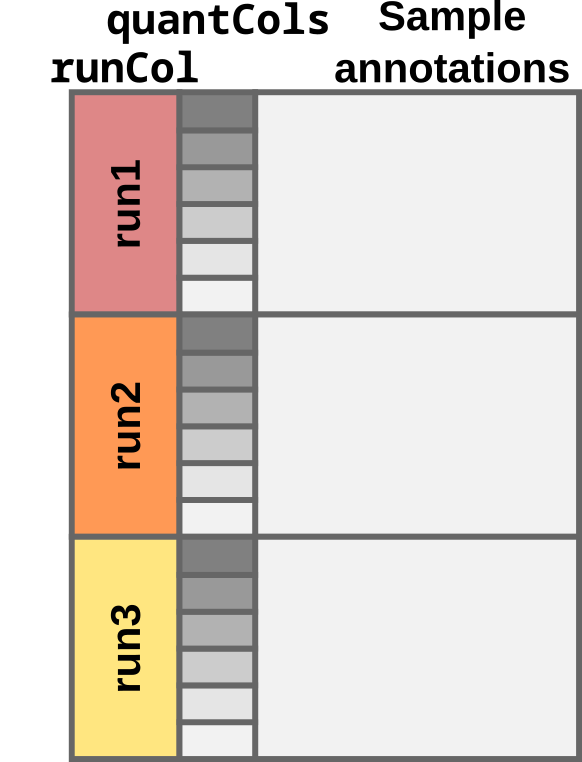
colData for the multi-set case
Let’s simulate an annotation table based on our previous example by duplicating the table for each run:
coldataMulti <- DataFrame()
for (run in paste0("run", 1:3, ".raw")) {
coldataMulti <- rbind(coldataMulti, DataFrame(runCol = run, coldata))
}
coldataMulti
#> DataFrame with 30 rows and 4 columns
#> runCol quantCols condition batch
#> <character> <integer> <character> <character>
#> 1 run1.raw 1 A batch1
#> 2 run1.raw 2 B batch1
#> 3 run1.raw 3 A batch1
#> 4 run1.raw 4 B batch1
#> 5 run1.raw 5 A batch1
#> ... ... ... ... ...
#> 26 run3.raw 6 B batch2
#> 27 run3.raw 7 A batch2
#> 28 run3.raw 8 B batch2
#> 29 run3.raw 9 A batch2
#> 30 run3.raw 10 B batch2We can provide the table to readQFeatures():
(qfMulti <- readQFeatures(
hlpsms, quantCols = quantCols, colData = coldataMulti,
runCol = "FileName"
))
#> Checking arguments.
#> Loading data as a 'SummarizedExperiment' object.
#> Splitting data in runs.
#> | | | 0% | |======================= | 33% | |=============================================== | 67% | |======================================================================| 100%
#> Formatting sample annotations (colData).
#> Formatting data as a 'QFeatures' object.
#> An instance of class QFeatures (type: bulk) with 3 sets:
#>
#> [1] run1.raw: SummarizedExperiment with 1004 rows and 10 columns
#> [2] run2.raw: SummarizedExperiment with 1004 rows and 10 columns
#> [3] run3.raw: SummarizedExperiment with 1002 rows and 10 columnsAdditional information
Sample names
readQFeatures() automatically assigns names that are
unique across all samples in all sets. In the single-set case, sample
names are provided by quantCols.
colnames(qfSingle)
#> CharacterList of length 1
#> [["quants"]] X126 X127C X127N X128C X128N X129C X129N X130C X130N X131In the multi-set case, sample names are the concatenation of the run
name and the quantCols (separated by a _).
colnames(qfMulti)
#> CharacterList of length 3
#> [["run1.raw"]] run1.raw_X126 run1.raw_X127C ... run1.raw_X130N run1.raw_X131
#> [["run2.raw"]] run2.raw_X126 run2.raw_X127C ... run2.raw_X130N run2.raw_X131
#> [["run3.raw"]] run3.raw_X126 run3.raw_X127C ... run3.raw_X130N run3.raw_X131Special case: empty samples
In some rare cases, it can be beneficial to remove samples where all
quantifications are NA. This can occur when the raw data
are searched for labels that were not used during the experiment. For
instance, some may quantifying the raw data expecting TMT-16 labelling
while the experiment used TMT-11 labels, or used half of the TMT-16
labels. The missing label channels are filled with NAs.
When setting removeEmptyCols = TRUE,
readQFeatures() automatically detects and removes columns
containing only NAs.
hlpsms$X126 <- NA
(qfNoEmptyCol <- readQFeatures(
hlpsms, quantCols = quantCols, removeEmptyCols = TRUE
))
#> Checking arguments.
#> Loading data as a 'SummarizedExperiment' object.
#> Formatting sample annotations (colData).
#> Formatting data as a 'QFeatures' object.
#> An instance of class QFeatures (type: bulk) with 1 set:
#>
#> [1] quants: SummarizedExperiment with 3010 rows and 9 columnsNote that we have set all values in X126 to missing.
Hence, the set contains only 9 columns instead of the previous 10.
Reducing verbose
Every call to readQFeatures() prints progression to the
console. To disable the console output, you can use the
verbose argument:
(qfSingle <- readQFeatures(
hlpsms, quantCols = quantCols, verbose = FALSE
))
#> An instance of class QFeatures (type: bulk) with 1 set:
#>
#> [1] quants: SummarizedExperiment with 3010 rows and 10 columnsUnder the hood
readQFeatures proceeds as follows:
- The
assayDatatable must be provided as adata.frame(or any format that can be coerced to adata.frame).readQFeatures()converts the table to aSingleCellExperimentobject usingquantColsto identify the quantitative values that are stored in theassayslot. Any other column is considered as feature annotation and will be stored asrowData.
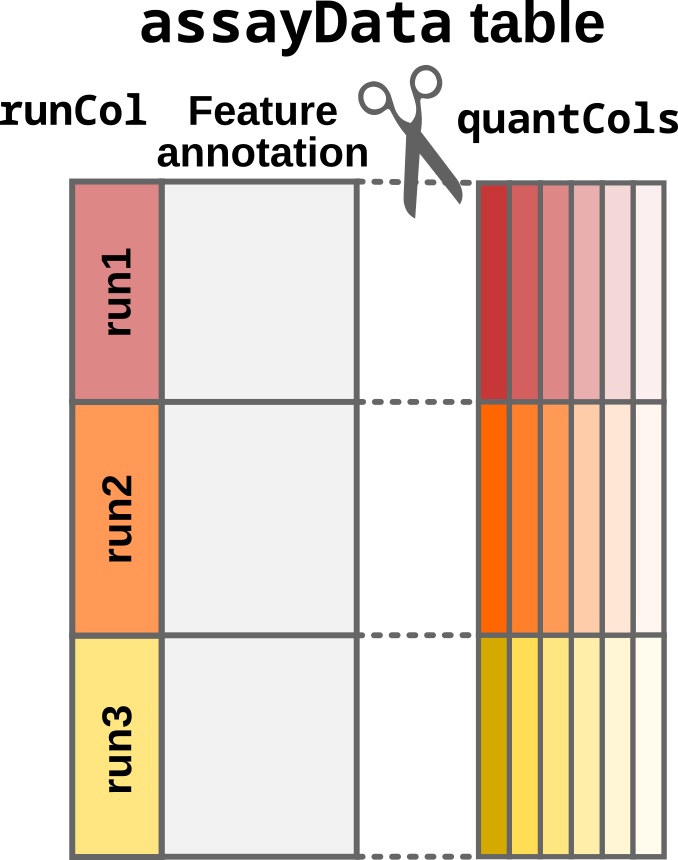
Step1: Convert the input table to a SingleCellExperiment
object
- (Only for the multi-set case:) The
SingleCellExperimentobject is split according to the acquisition run provided by therunColcolumn inassayData.
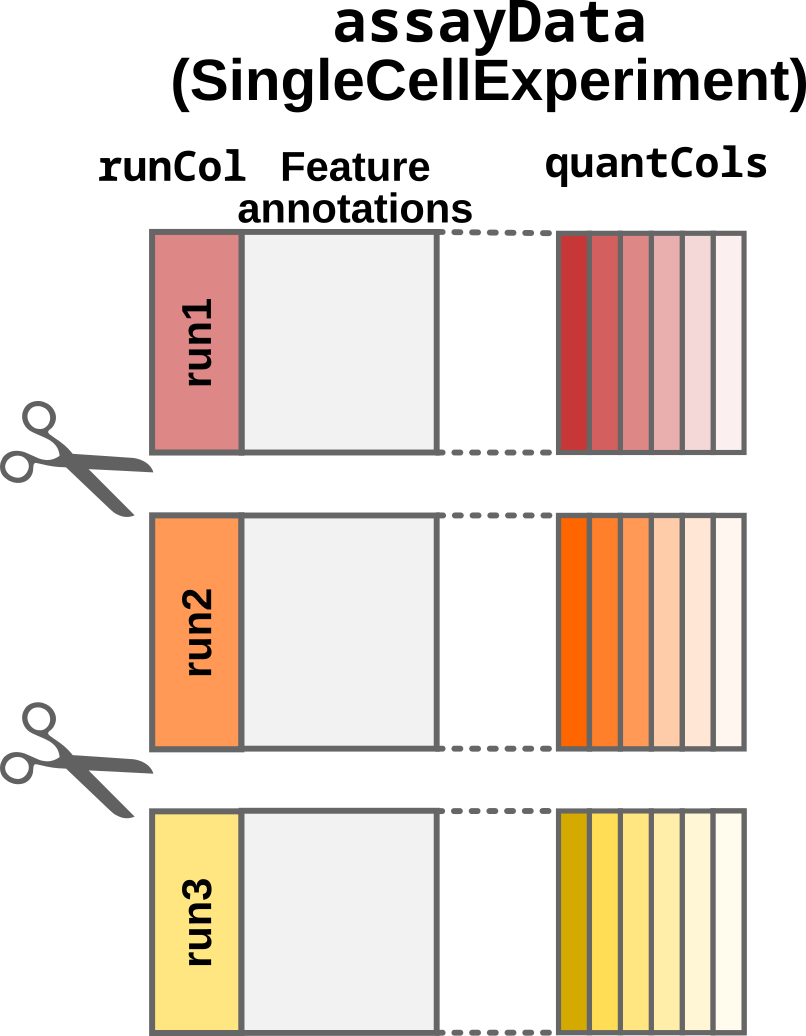
Step2: Split by acquisition run
- The sample annotations are generated. If no
colDatais provided, the sample annotations are empty. Otherwise,readQFeatures()matches the information fromassayDataandcolDatabased onquantCols(single-set case) orquantColsandrunCol(multi-set case). Sample annotations are stored in thecolDataslot of theQFeaturesobject.
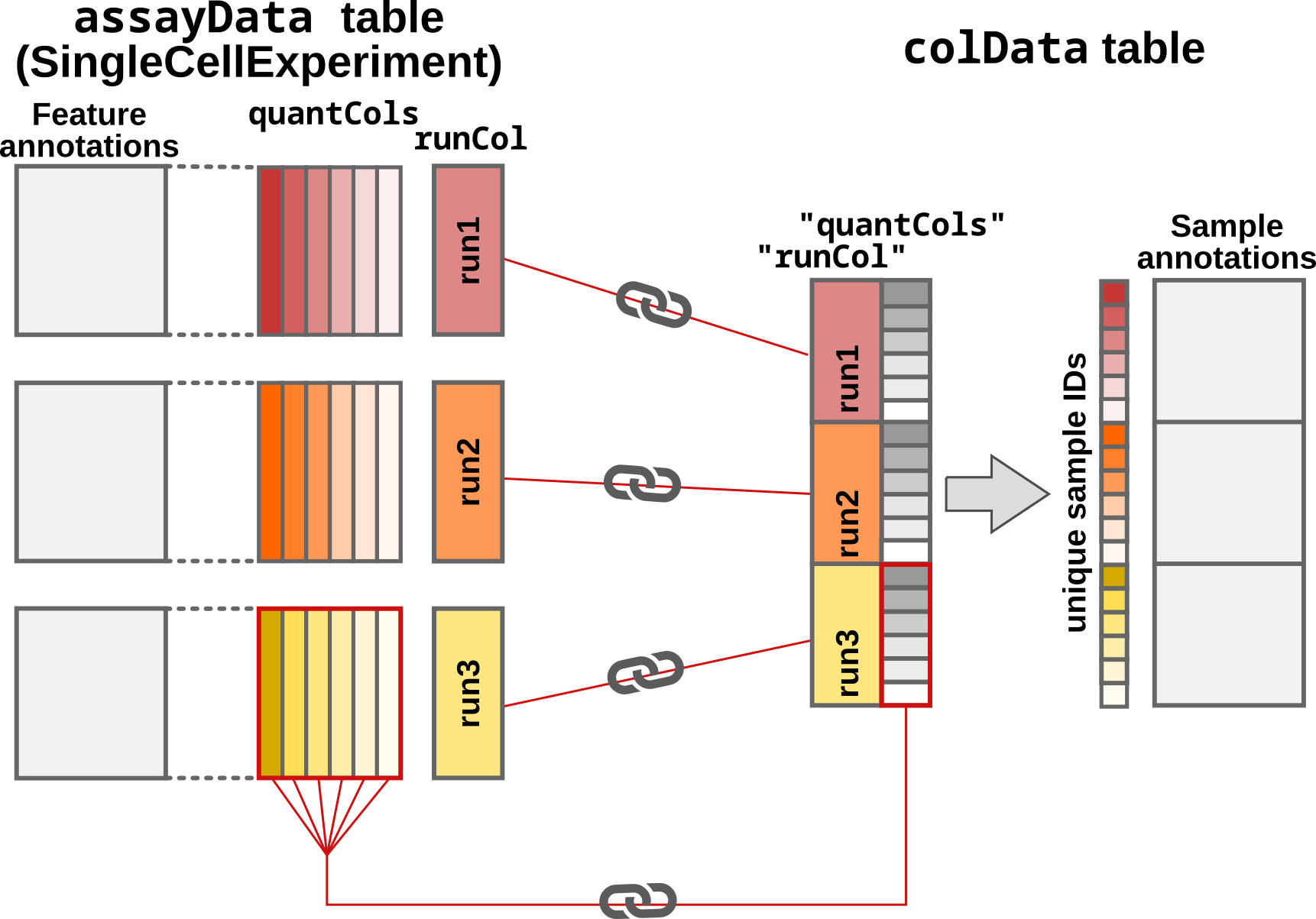
Step3: Adding and matching the sample annotations
- Finally, the
SummarizedExperimentsets and thecolDataare converted to aQFeaturesobject.
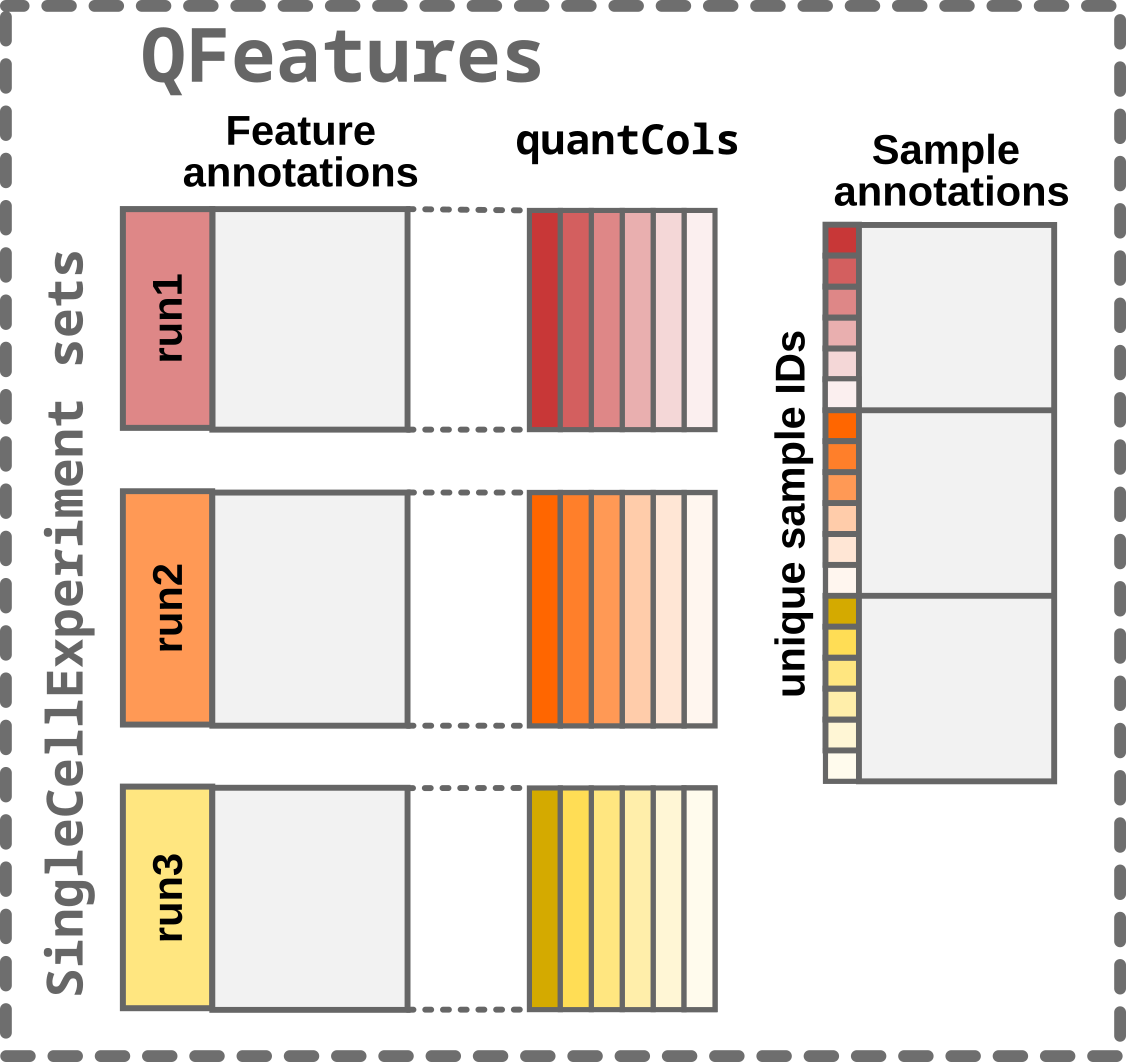
Step4: Converting to a QFeatures
What about other input formats?
readQFeatures() should work with any PSM quantification
table that is output by a pre-processing software. For instance, you can
easily import the PSM tables generated by Proteome Discoverer. The run
names are contained in the File ID column (that should be
supplied as the runCol argument to
readQFeatures()). The quantification columns are contained
in the columns starting with Abundance, eventually followed
by a multiplexing tag name. These columns should be stored in a
dedicated column in the colData data to be supplied as
runCol to readQFeatures().
The QFeatures package is meant for both label-free and
multiplexed proteomics data. Importing LFQ data is similar to the
examples above with the only difference that quantCols
would have only 1 element.
The readSCPfromDIANN() function is adapted to import
label-free and plexDIA/mTRAQ Report.tsv files generated by
DIA-NN.
For more information, see the ?readQFeatures() and
?readQFeaturesFromDIANN() manual pages, that described the
main principle that concern the data import and formatting.
If your input cannot be loaded using the procedure described in this vignette, you can submit a feature request (see next section).
Need help?
You can open an issue on the GitHub
repository in case of troubles when loading your data with
readQFeatures(). Any suggestion or feature request about
the function or the documentation are also warmly welcome.
Session information
R Under development (unstable) (2026-04-12 r89873)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 24.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: UTC
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] DT_0.34.0 QFeatures_1.21.2
[3] MultiAssayExperiment_1.37.4 SummarizedExperiment_1.41.1
[5] Biobase_2.71.0 GenomicRanges_1.63.2
[7] Seqinfo_1.1.0 IRanges_2.45.0
[9] S4Vectors_0.49.1 BiocGenerics_0.57.0
[11] generics_0.1.4 MatrixGenerics_1.23.0
[13] matrixStats_1.5.0 BiocStyle_2.39.0
loaded via a namespace (and not attached):
[1] xfun_0.57 bslib_0.10.0 htmlwidgets_1.6.4
[4] lattice_0.22-9 crosstalk_1.2.2 vctrs_0.7.3
[7] tools_4.7.0 tibble_3.3.1 cluster_2.1.8.2
[10] pkgconfig_2.0.3 Matrix_1.7-5 desc_1.4.3
[13] lifecycle_1.0.5 compiler_4.7.0 stringr_1.6.0
[16] textshaping_1.0.5 clue_0.3-68 htmltools_0.5.9
[19] sass_0.4.10 yaml_2.3.12 lazyeval_0.2.3
[22] pkgdown_2.2.0.9000 pillar_1.11.1 jquerylib_0.1.4
[25] tidyr_1.3.2 MASS_7.3-65 DelayedArray_0.37.1
[28] cachem_1.1.0 abind_1.4-8 tidyselect_1.2.1
[31] digest_0.6.39 stringi_1.8.7 purrr_1.2.2
[34] dplyr_1.2.1 reshape2_1.4.5 bookdown_0.46
[37] fastmap_1.2.0 grid_4.7.0 cli_3.6.6
[40] SparseArray_1.11.13 magrittr_2.0.5 S4Arrays_1.11.1
[43] rmarkdown_2.31 XVector_0.51.0 igraph_2.2.3
[46] otel_0.2.0 ragg_1.5.2 evaluate_1.0.5
[49] knitr_1.51 rlang_1.2.0 Rcpp_1.1.1
[52] glue_1.8.0 BiocManager_1.30.27 jsonlite_2.0.0
[55] AnnotationFilter_1.35.0 R6_2.6.1 plyr_1.8.9
[58] systemfonts_1.3.2 fs_2.0.1 ProtGenerics_1.43.0
[61] MsCoreUtils_1.23.7 License
This vignette is distributed under a CC BY-SA license license.